Ex64 吸附能的计算(九)

前面我们讲解了一堆单原子的吸附计算和一些日常的操作,现在是时候来点复杂的操练了。这次,我们拿CO开刷,计算它在Cu(111)表面上的吸附。由于CO比前面的O多了一个原子,复杂性稍微提高了些,但也不是太复杂。作为一个由简单向复杂体系的过渡,是一个很好地例子。
在计算CO的吸附之前,我们首先要了解以下3点:(都是教科书里面的经典内容,下面只列出来,不再细说,如果不懂的话,找本结构化学书好好啃一啃。)
1. CO分子信息:
1)CO的分子结构,如下图:
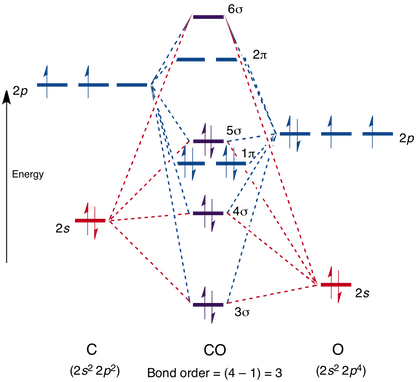
2) CO的几何结构:(用来搭建吸附和气相的模型)

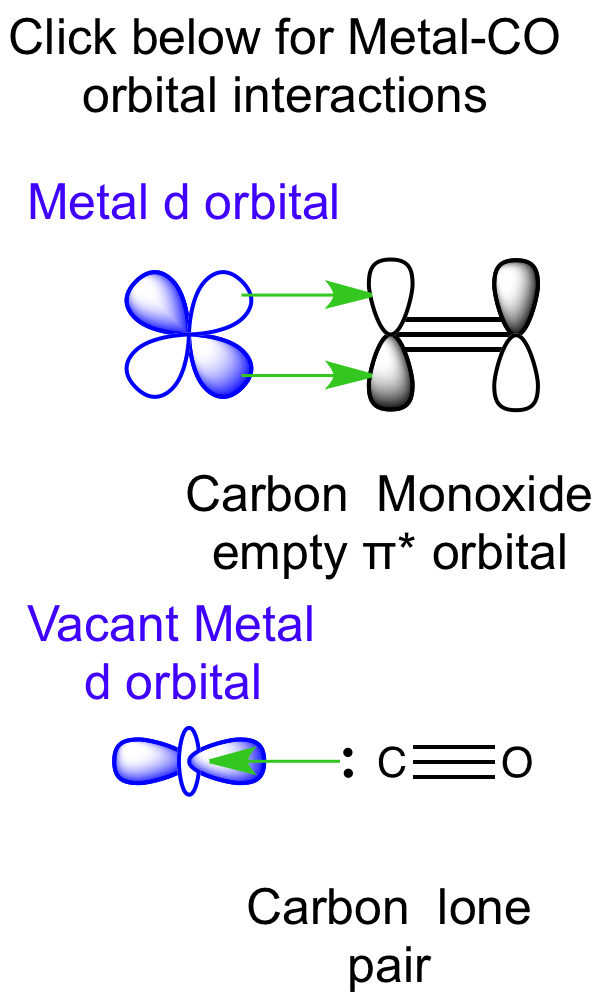
3) CO与金属的成键方式 (用来搭建吸附模型)
2. 搭建CO吸附的模型:
上一节我们学到了闭着眼操作的一些方式,这一节我们继续闭着眼搭结构。操作流程如下:
1) 将上一节中 C 原子吸附的那几个模型复制过来,进行修改:
- i) 修改POSCAR中的元素(第6行)和原子数目部分(第七行),添加O原子。
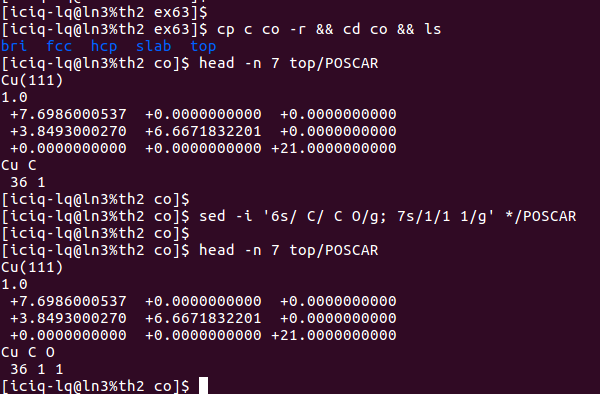
- iii)在POSCAR的尾部,添加O原子的坐标:
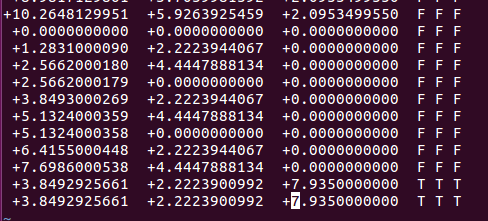
可以看到上图中,最后两行的坐标一模一样,这是因为CO在表面上吸附的时候,我们假定的是直立吸附。所以C 和 O原子的坐标在x和y方向上一样。不同的区别在z方向上,即CO的键长。CO如果吸附在表面上,肯定会和表面原子有作用,也就是所谓的活化,如果原子被活化了,那么CO键就会被削弱,具体体现在键长上。与气相的键长相比,表面上CO的键长数值更大一些。前面我们查数据库得到CO键长为:1.138A.这里我们不妨设置成1.2A。根据这些,我们设置O原子的坐标,如下:
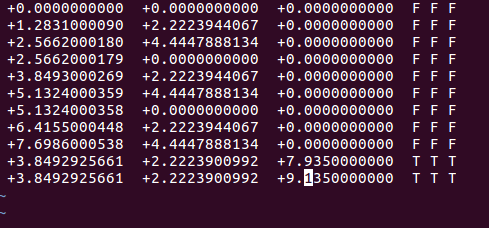
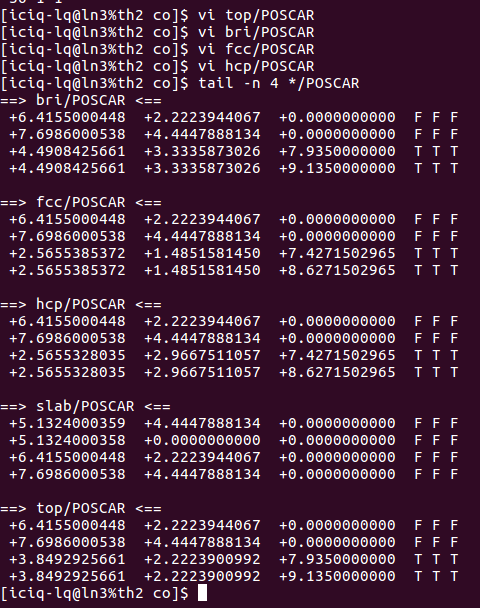
同样,我们可以对其他吸附位点的坐标进行类似的修改,结果如下图:
3. 背一遍VASP的输入文件,检查还有那些需要修改的:
- i) INCAR: 跟之前保持一致;
- ii)KPOINTS:跟之前保持一致;
- iii) POSCAR: 已经修改完毕;
- iV) POTCAR:提交任务的脚本里面自动生成
- V)提交任务命令: qsuball (前面已经讲解过了)
4. 提交任务:
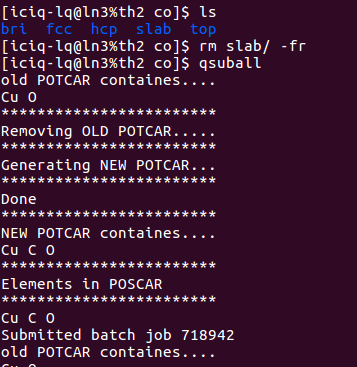
上图,我们删除了slab的计算,因为前面我们已经计算过了,没有必要浪费机时再算一遍。
5. 思考下吸附能的计算公式:
前面我们还忘记了CO的气相结构优化。现在我们回忆一下前面所讲的气相分子的优化:
1) 气相分子的结构模型搭建。
直接将一个Cu(111)表面上吸附的POSCAR拿过来修改一下即可:
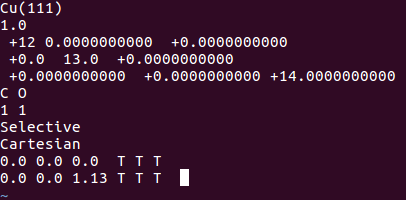
2) INCAR:直接拿Cu(111)吸附的修改下。
对于气相分子的优化来说:
1 | ISMEAR = 0 |
3) KPOINTS: gamma 点即可。
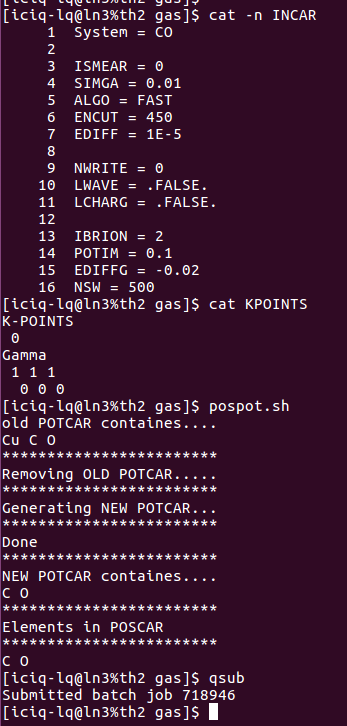
4) 这里我们没有批量提交,手动运行一下:pospot.sh脚本,生成对应的POTCAR。
5) 使用提交单个任务的脚本: qsub 提交任务。具体操作如下图:
6. 扩展练习:
- 1)完成CO吸附的计算;
- 2)进一步熟练简单模型的闭着眼操作方式;
- 3)复习分子气相结构的优化过程。
7. 总结:
本节,通过CO的吸附模型搭建,带领大家走出简单的单原子吸附,开始逐渐接触复杂的吸附计算。如果前面内容掌握了,本节就是一个水到渠成的事情。在多原子分子的吸附计算中,首先我们要知道分子的电子和几何结构,分子哪一部分(这里的C原子)和表面成键。在本节的例子中,如果你不知道C和金属作用,你还需要计算Metal-O-C这样的结构。可能还会计算CO横着吸附的结构,任务无形中就会增加一倍或者更多,从而造成机时的浪费。退一步来说,如果你真的不知道吸附是以什么方式进行的,想尝试N种初始的结构,我的建议是:把slab的原子全部固定住,然后用gamma点算一下它们的吸附能,先大体上判断一下,把那些吸附特别强的结构筛选出来,用作下一步的计算。