Ex65 p4vasp 的旋转操作
经过前面的学习,相信对于:单原子和双原子分子的吸附,大家已经掌握了怎么搭建模型,以及计算吸附能了。下一节我们要讲的是三原子分子的吸附。在进行三原子分子吸附的计算之前,我想是时候祭出本人使用p4vasp搭结构的一个法宝了。古人语,工欲善其事,必先利其器。所以,本人认为这一节的学习对于以后的结构搭建非常重要,尤其是对于那些使用p4vasp的筒子们来说。说了这么多废话,本节主要内容是手动搭建一个H$_2$O分子,并实现分子的任意旋转。
1. 获取结构
首先我们先知道H$_2$O分子的基本结构。本人的做法是去NIST数据库查找资料。操作如下图:
点击上的链接,或者点击 NIST网址查询得到下面的信息:
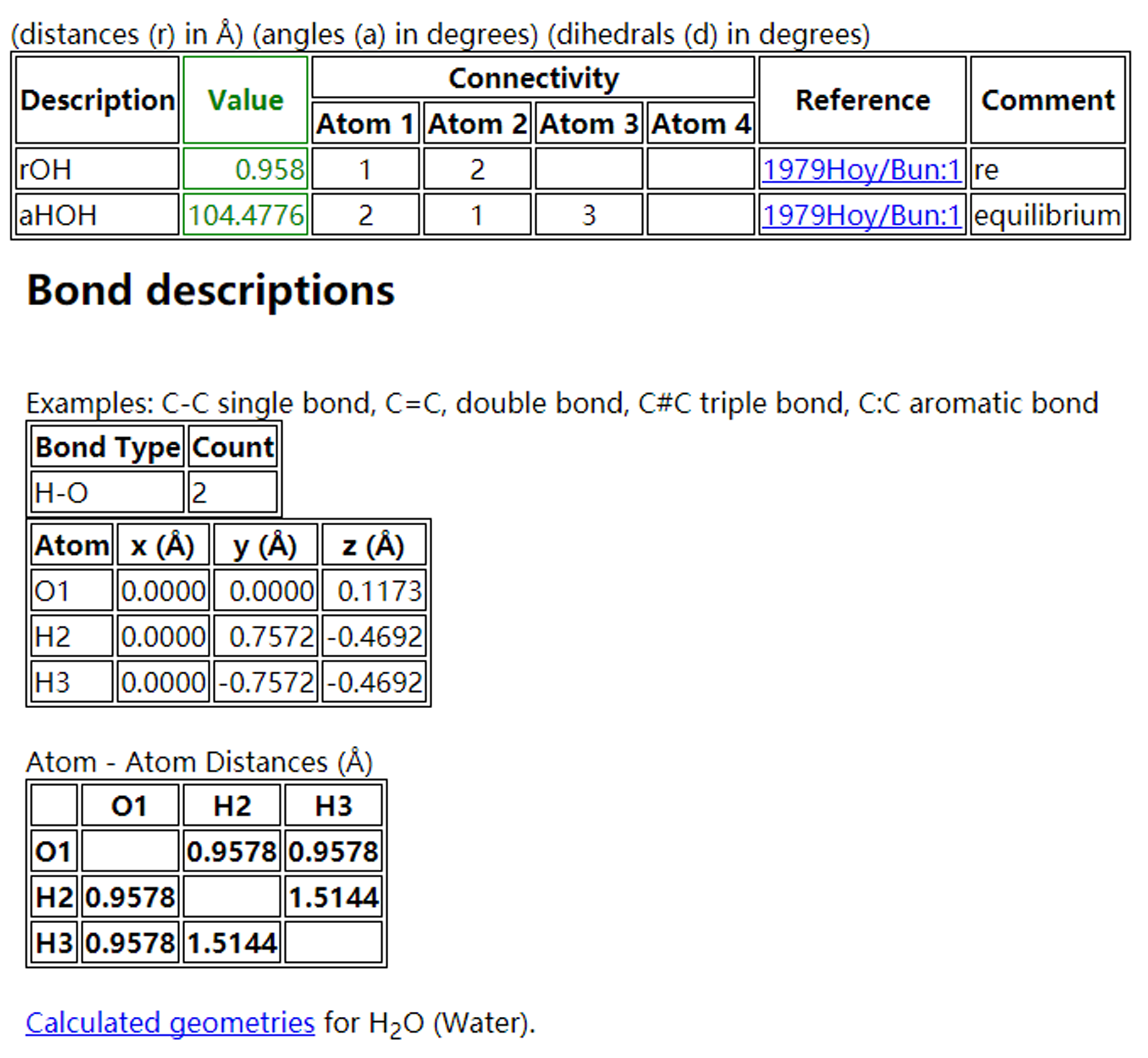
在NIST的数据库里面,基本的热力学数据,结构都有了。大家完全可以按照上图中的Cartesian坐标搭建一个H$_2$O的结构,直接复制到POSCAR即可。不过,这个操作我们先缓一下,以后你有的是机会操作。本节主要介绍下如何是从头开始搭建一个H$_2$O分子模型,整个操作过程的学习比搭建模型的结果更加重要。
2. 模型搭建:
1)数据库结构
O—H的键长为0.9578 A,∠HOH = 104.478°。
2)直角形H2O分子
首先,我们在xy平面内搭建一个直角形的H$_2$O分子。O放到原点: 0, 0, 0,H_1沿着x轴, 0.96, 0, 0,H_2 沿着y轴: 0, 0.96, 0
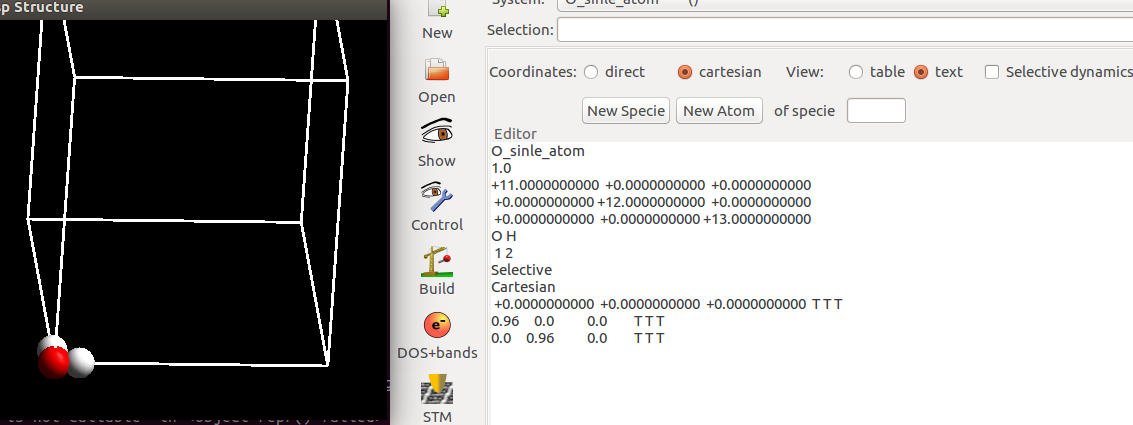
3)修改角度
前面说了,∠HOH = 104.478°, 我们该怎么做呢?
i) 可以自己手动算一算,其中一个H原子的坐标,然后更新坐标信息。三角公式大师兄早就忘的一干二净了,暂时跳过。
ii)通过p4vasp进行旋转操作。
对于旋转操作,我们首先要定义一个旋转轴;然后选择旋转的原子,以及角度。在上面H$_2$O的结构里面,很容易就想到,如果∠HOH 从 90°增大到 104.478°, 我们需要以O原子所在的z轴,旋转一个H原子即可,旋转角度为14.478°。
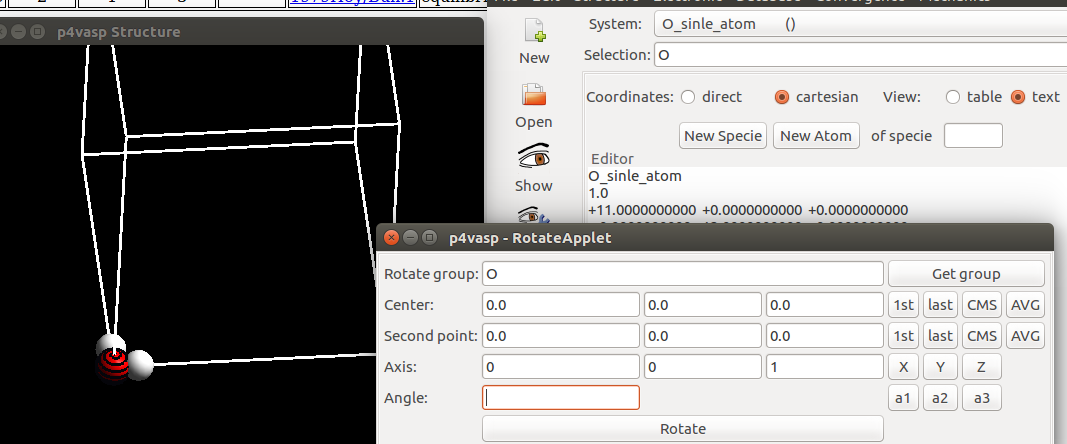
4) p4vasp获取旋转轴的操作:
- i) 选中O原子;
- ii) P4vasp界面左上方,
Edit—>Rotate Atoms; - iii) 弹出的界面点击:
Get Group; - iv)
Center那一行,点击1st的按钮; - v)
Second point那一行,点击1st的按钮; - vi)
Axis那一行,点击Z的按钮。
这样我们就定义了一个穿过O原子的z轴。
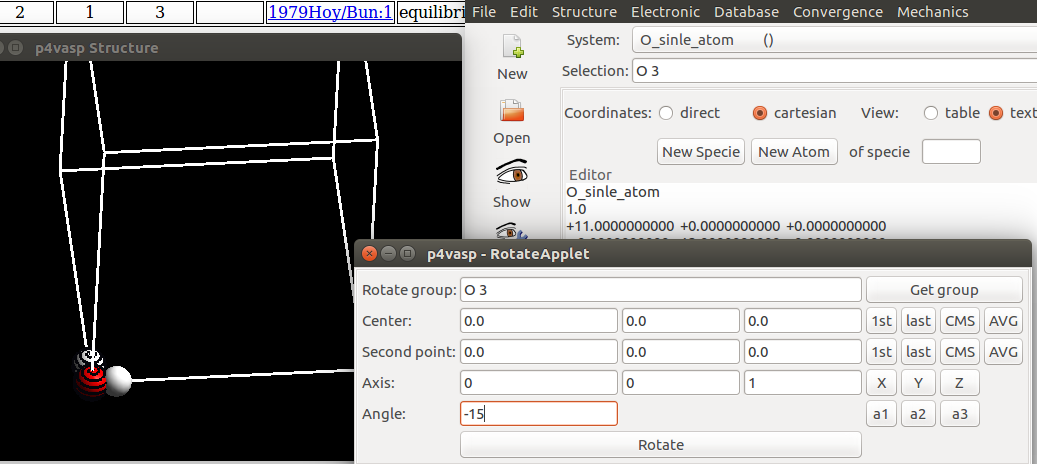
5) p4vasp选择要旋转的原子和角度:
- i)把鼠标分别移动要要旋转的H上,通过键盘的
space空格键选择。 - ii)在上一步
Edit–>Rotate Atoms弹出的对话框中,再点一下:Get Group。 - iii)最后一行,我们选择 -15 作为旋转的角度。
注意1: +15 和 -15 顺时针和逆时针旋转15°。 如果你旋转错了,不要心慌,把数值改下,重新旋转即可。
注意2: 你也可以先选中O和H原子,然后在center 和 second point 那两行都选择1st来获取旋转轴。
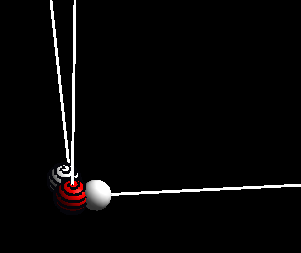
点击最下面的: Rotate,效果如下:
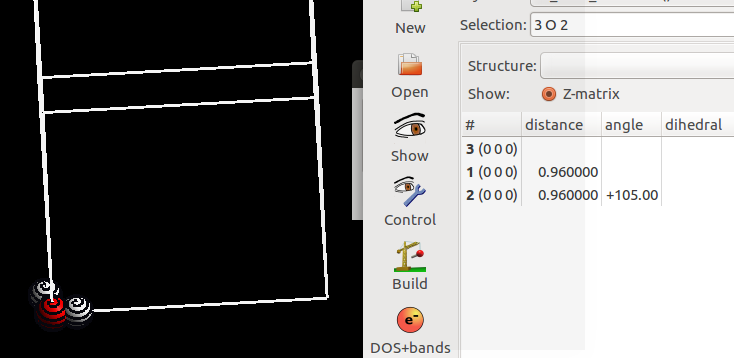
由于我们绕着O原子旋转,所以O原子选中或者不选中,旋转操作都对其坐标都没有影响。通过Structure – Measure 测定一下 三个原子的角度。
3. 绕任意轴旋转
上面我们介绍了一下,绕一个单原子的轴进行的旋转操作。而实际的模型调整,搭建的过程中,这种情况并不多,大多时候我们需要绕着一个3D空间里面的轴进行旋转,而不仅仅局限在xyz这样简单的情况。我们知道,两点可以确定一条直线,所以,对于旋转轴来说,我们可以通过两个原子来定义。下面我们讲解一下,H$_2$O分子绕着一个O—H键的旋转操作。
1)定义旋转轴:
- i) 选择 O 原子 和 H原子;
- ii) Edit –-> Rotate Atoms;
- iii) Get group;
- iv) Center 那一行,点 1st;
- v) Second point那一行,点 last。
点完之后,下面一行会自动填充我们选择的旋转轴。即从1st原子指向last原子的一个轴。最后一部分,我们写的是60,即绕着O—H键 旋转60°。
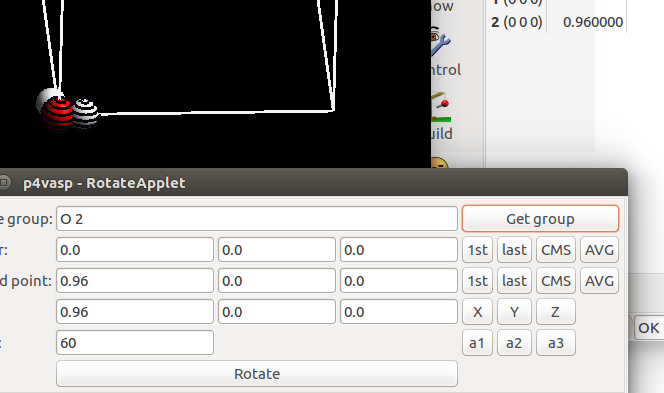
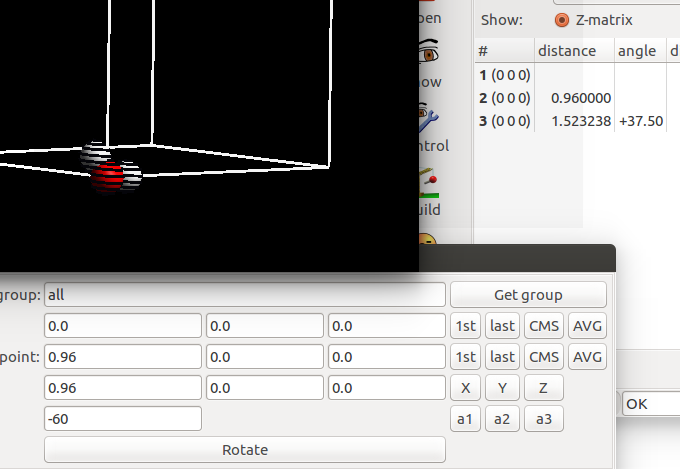
选择要旋转的原子,由于前面两个原子已经用来定义旋转轴了,剩下的第三个就是我们旋转操作的对象。选中所有的原子,然后点击 Get group。 下面的图是:H$_2$O 的一个H原子绕着另外两个原子的轴旋转 -60 °的效果。我们绕着一个O—H 键旋转,旋转操作对轴上的原子坐标没有影响,所以大家可以选中这些原子,也可以先通过这两个原子定义旋转轴,进行最后一步操作的时候,取消选择也可以。
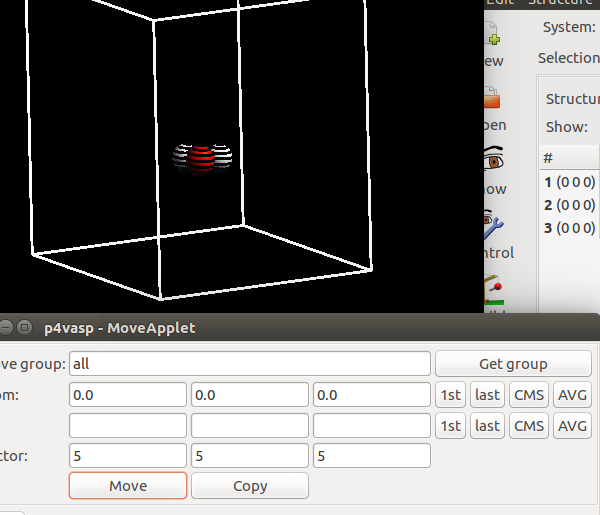
2)移动水分子
一般对于分子的计算来说,如果分子在原点附近的话,由于周期性的原因,结构的一部分会进入到另外一个相邻的格子里面,虽然对于计算没有什么影响,但对后面的其他可视化过程(比如,频率计算等)会造成一定的影响。所以本人经常把原子移到格子里面。选择所有的原子,在最后的Vector部分,选择5 5 5, 即在xyz三个方向上都移动5A的距离。点击move,效果如下图,然后保存结构。
4. 扩展练习:
- 1) 重复本节的所有操作;
- 2) 复习前面乙醇分子模型的搭建
- 3) 根据今天所学,随意操作乙醇分子中,原子的旋转,平移等操作,直至熟练位置。
5. 总结:
本节的重点是学会用p4vasp 进行分子的旋转操作,并复习下分子的平移操作。分子的旋转在表面结构模型的搭建过程中,非常重要,熟练掌握这一个技巧,可以极大提高自己搭建模型的合理性和准确性,从而在后面的计算过程中,节约我们的计算时间。如果你有更好的方法,也也可以分享经验给大家。