Ex-37 DOS 计算(一)
使用VASP计算,很多时候都逃不掉DOS,能带计算的相关问题,尤其是对于计算材料的童鞋们,更是家常便饭一般。群里很多人,很多新手们都时常在讨论DOS的计算。这里我们通过VASP官网的说明,解释一下算DOS的具体步骤。前面我们学会了如何拟合或者优化稳定的晶胞结构。在此基础上,我们可以计算一下相关的DOS信息。
1 KPOINTS
1.1 K点数目
与结构优化相比,算DOS的时候,需要用到更多的K点数目,这是因为K点越多,画出来的DOS图质量越高。
引用官网的话:
1 | A high quality DOS requires usually very fine k-meshes. |
1.2 K点数目的选取
K点数目越多越好,我们该如何设置K点数目呢?
还记的前面我们讲到的K点选择的经验规则吗?那一个规则可以认为是我们平时计算时K点选择的标配。对于DOS计算,我们就需要把配置提高一个档次了。

一般来说,K * a = 45左右之间完全可以满足你的要求,大伙可以根据这个经验来选择K点。
2 NEDOS
NEDOS这个参数在DOS图的质量上面也有着很重要的作用。比如我们的DOS能量区间范围(DOS图的横坐标)为:[-10 eV,10eV],VASP默认的将这个能量范围分成301点,然后作图。301也就是默认的NEDOS的取值。如果我们设置的NEDOS值够大,那么DOS区间就会被区分地越精确。NEDOS的取值一般来说:
NEDOS = 3000左右就足够好了。太大也没什么意义;NEDOS越大,VASP输出的DOSCAR,vasprun.xml文件也就越大,占用存储空间。
经常有人抱怨说自己的DOS图有很多尖锐的峰,可以尝试着通过增加NEDOS这个办法来解决。
更多的信息,自己参考一下:https://cms.mpi.univie.ac.at/wiki/index.php/NEDOS
3 ISMEAR(一)
首先我们看官网的话: https://cms.mpi.univie.ac.at/wiki/index.php/ISMEAR
1 | For the calculation of the total energy in bulk materials we recommend thetetrahedron method with Blöchl corrections (ISMEAR=-5). This method also givesa smooth nice electronic density of states (DOS). |
也就是说 ISMEAR = -5 的时候(Blöchl修正的四面体方法),我们可以得到一个非常平滑的DOS图。
注意:
3.1 K点数目
设置ISMEAR = -5 的时候,如果K点数目K点的数目小于等于4 , 计算会出错,得到如下的错误结果:
VERY BAD NEWS! internal error in subroutineIBZKPT:
Tetrahedron method fails for NKPT<4. NKPT= 1
这也是很多人常见的错误。官网说的是K点数目小于三:the tetrahedron method is not applicable, if less than three k-points are used. (QQ群的恒驰一强发现官网的这个错误。)
K点不够,用ISMEAR = -5出错的解决办法:
既然K点不够,那么我就增加K点,然后再使用
ISMEAR= -5(简单粗暴,强烈推荐使用)如果增加了K点,可能还是会出错。有时也会出现下面的错误(微信群的群友(Cu—Ni)提供)。我们先把解决方法列出来,错误部分大家慢慢看:
- 直接换一下ISMEAR的取值。
- 群友还发现:在保证K点数目大于4的时候,有时候减少K点数目或者增加K点数目都可以解决这个问题。如果你的服务器还算可以,建议增加K点数目,毕竟和K点数目越多,DOS的质量越高。这个办法大家可以参考一下。
1
2WARNING: DENTET:can't reach specified precision
Number of Electronsis NELECT =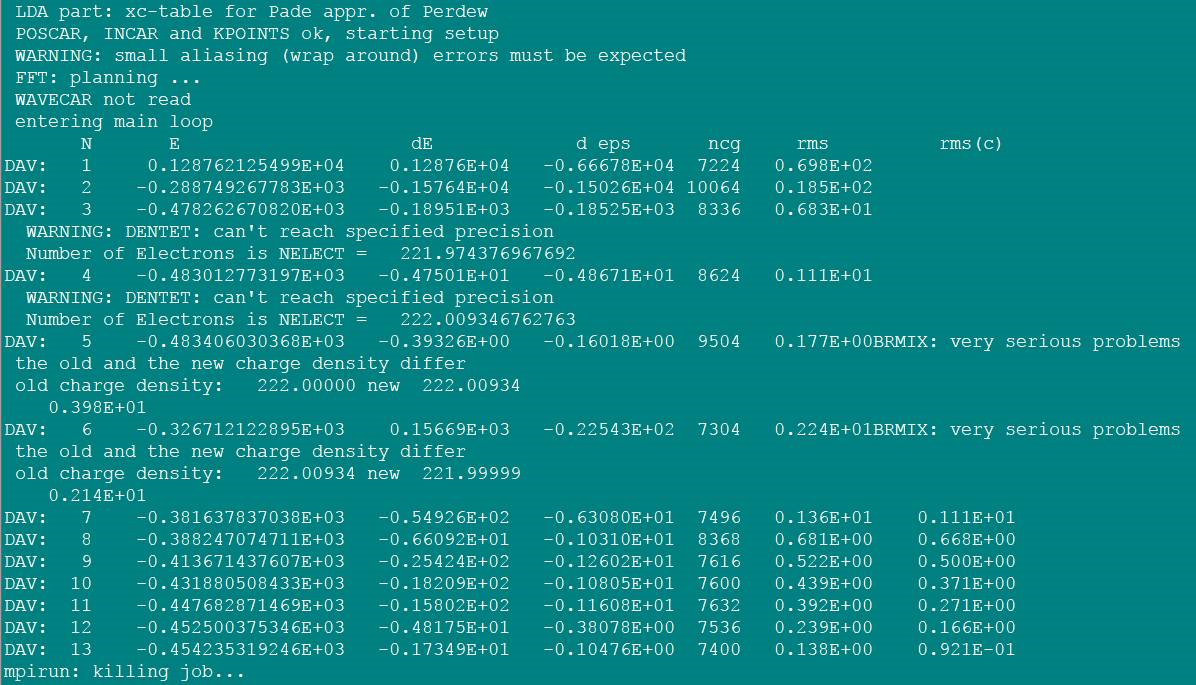
官方的解释:
http://cms.mpi.univie.ac.at/vasp-forum/viewtopic.php?t=416
http://www.error.wiki/The_old_and_the_new_charge_density_differ

出现此警告(DENTET)的原因是因为无法通过tetrahedron方法得到足够精确的费米能级。也就是将态密度积分到费米面的电子数和体系的价电子数目不一致。
3.2 适用体系:
ISMEAR = -5适用于所有体系的DOS计算。非DOS计算的时候:对于金属体系来说,结构优化的时候不能使用
ISMEAR= -5(注意:是优化结构的时候不能用!),这是因为四面体方法不能很好地处理费米能级处的电子占据情况,导致算出来的力会有一定百分比的误差。所以,对金属体系结构优化的时候,ISMEAR > = 0参考:https://cms.mpi.univie.ac.at/vasp/vasp/Partial_occupancies_different_methods.html对于半导体和绝缘体的体系,
ISMEAR>0是不可以的。只能ISMEAR<=0。四面体方法(
ISMEAR = -5)不适合计算能带(对所有的体系来说的)。多谢wuli8老师帮忙完善!- 使用ISMEAR= -5 的时候,SIGMA的取值没有影响,如果不放心,取
SIGMA = 0.01。
3.3 小结:
- 算DOS,只要K点不少于3; 用ISMEAR=-5是个不错的选择。
4 ISMEAR(二)
如果体系很大,只能适用gamma点来算,ISMEAR = -5的时候,肯定会出错,但服务器不给力,不能增加K点的时候,怎么办?
对于所有的体系(K点数目小于4也可以):可以使用
ISMEAR = 0;SIGMA = 0.01。对于大部分的体系都能得到理想的结果。原则上来说,使用GS方法的时候(ISMEAR=0),SIGMA的数值要测试下,保证entropy T*S这一项平均到每个原子上小于0.001 eV也就是1meV。不想测试的话,直接用个很小的值,比如这里我们说的:SIGMA = 0.01。
对于金属体系来说,也可以使用
ISMEAR = 1;SIGMA = 0.01。SIGMA取值太大,计算出来的能量可能不正确;SIGMA取值越小,计算越精确,需要的时间也就越多。值得注意的是:这里我们使用的0.01已经很小,没有必要设置的再小。因为对于金属体系,使用MP方法(ISMEAR=1..N)时,SIGMA= 0.10 差不多就足够了。官网给的参考值是0.20。
http://cms.mpi.univie.ac.at/vasp/guide/node124.html

- 在KPOINTS确定之后,使用多大的SIGMA值,大家最好测试一下。原则如下:SIGMA取值在保证OUTCAR中
entropy T*S这项的能量平均到每个原子上小于 1 meV的前提下:尽可能地大。这样可以在保证准确度的同时,也加快收敛速度。

- 记住: VASP学习最快的途径就是不停地看官网,然后亲自上手去测试,测试,测试!并观察分析结果。
4 扩展练习:
4.1 阅读VASP官网关于ISMEAR和SIGMA的所有说明:
4.2 下载VASP的pdf说明书,搜索书中所有的ISMEAR和SIGMA关键词,阅读所有相关的内容;
4.3 思考SMEAR方法的意义?SIGMA的意义?
4.4 查看VASP说明书,查阅相关文献,了解MP和GS方法
4.5 测试MP和 GS方法中,SIGMA取值对计算时间,能量,收敛步数的影响。
4.6 分析下为什么算DOS的时候,要算两步: selfconsistent 和 none-selfconsistent calculations?
5 总结:
看完本节:你应该知道计算DOS的时候:
- KPOINTS和NEDOS设置的一些内容。
- ISMEAR要用-5。
KPOINTS因计算硬件限制不能设置的很大,数目小于4的时候:
- 对于所有体系均可以使用ISMEAR=0。
- 金属体系还可以用ISMEAR=1..N,官网建议SIGMA为0.20,太小的SIGMA值对收敛会产生影响。使用0.01-0.10的数值都是很安全的选择。
- SIGMA的数值需要测试一下,一般来说在0.01-0.05之间足够了。
非DOS计算的时候,对于金属来说ISMEAR不能等于 -5,优先使用ISMEAR= 1。非金属来说(半导体和绝缘体),不能 > 0 。对于所有的体系, ISMEAR= 0 则是一个很安全的选择,但SIGMA的数值要测试一下。说了这么多废话,还是官网简单明了:

For further considerations on the choice for the smearing method see sections 9.4, 10.6. To summarize, use the following guidelines:
- For semiconductors or insulators use always tetrahedron method (ISMEAR=-5), if the cell is too large (or if you use only 1 or two k-points) use ISMEAR=0.
- For relaxations in metals always use ISMEAR=1 and an appropriated SIGMA value (the entropy term should less than 1 meV per atom). Mind: Avoid to use ISMEAR>0 for semiconductors and insulators, it might result in problems.
For metals a sensible value is usually SIGMA= 0.2 (that’s the value we use for most transition metal surfaces).
- For the DOS and very accurate total energy calculations (no relaxation in metals) use the tetrahedron method (ISMEAR=-5).