Ex72 过渡态的计算(三)
不好意思,让大家久等了。最近一直很忙,后面还是会很忙。为了不让大家失望,抽时间把简单的过渡态计算讲解一下,这一节我们需要学习的是通过CI-NEB(Climbing Image Nudged Elastic Band)计算H原子在Ru(0001)表面上的扩散过程。
NEB计算过渡态的准备工作
准备工作1:
说到NEB或者CINEB,不得不提的就是Henkelman课题组的VTST:http://henkelmanlab.org/ . 我们需要下载VTST的code,然后将VASP编译一下,得到一个可以使用vtst的VASP版本。
链接:http://theory.cm.utexas.edu/vtsttools/download.html
很不幸,怎么编译,我一窍不通。如果这一关过不了的话,后面的也只能看看热闹,不能亲自实践了。如果你已经编译好了VTST的VASP版本, 那么就可以继续下面的学习了。
准备工作2:
我们将VTST的一些实用小脚本下载下来:http://theory.cm.utexas.edu/vtsttools/download.html
下载VTST Scripts后解压,把所有脚本都复制到 ~/bin 目录下:效果如下:
很多脚本估计你一辈子都不会用到,没关系,它们很小,不占存储空间,静悄悄躺在bin文件里面就行了。Linux老手们嫌烦可以把这些脚本放到一个文件夹里面,然后bashrc文件里面修改下路径就可以了。
准备工作3:
修改nebresults.pl 文件,将57到71行注释掉,注释掉就是在每一行的开头加个#号,效果如下:
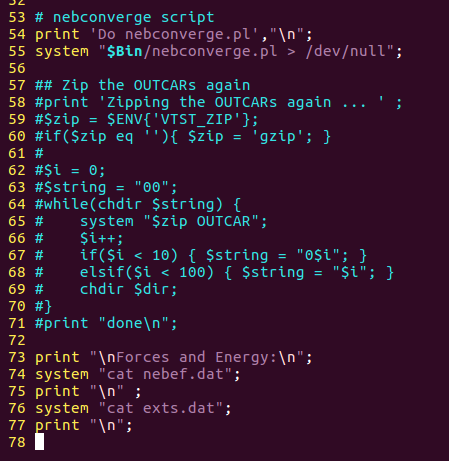
为什么需要这么做?
如果neb运行的时候,你使用这个命令,它就会把你的OUTCAR文件压缩,VASP找不到OUTCAR,就不知道该往哪里存储,然后就挂掉了。大家可以在neb任务运行的时候分别运行下注释前后的脚本,对比下就清楚了。
扩散的基础知识
准备工作完成,我们加深下扩散(diffusion)的一些概念:
2.1 扩散我们高中的时候应该就学习过了。具体的定义这里不讲了。大家自行参考教科书或者维基百科:链接如下:
https://en.wikipedia.org/wiki/Diffusion
对于分子在表面上的扩散,就跟我们在校园里瞎晃悠是一样的。哪天指不定遇到个对上眼的美女,过渡态就要开始了。
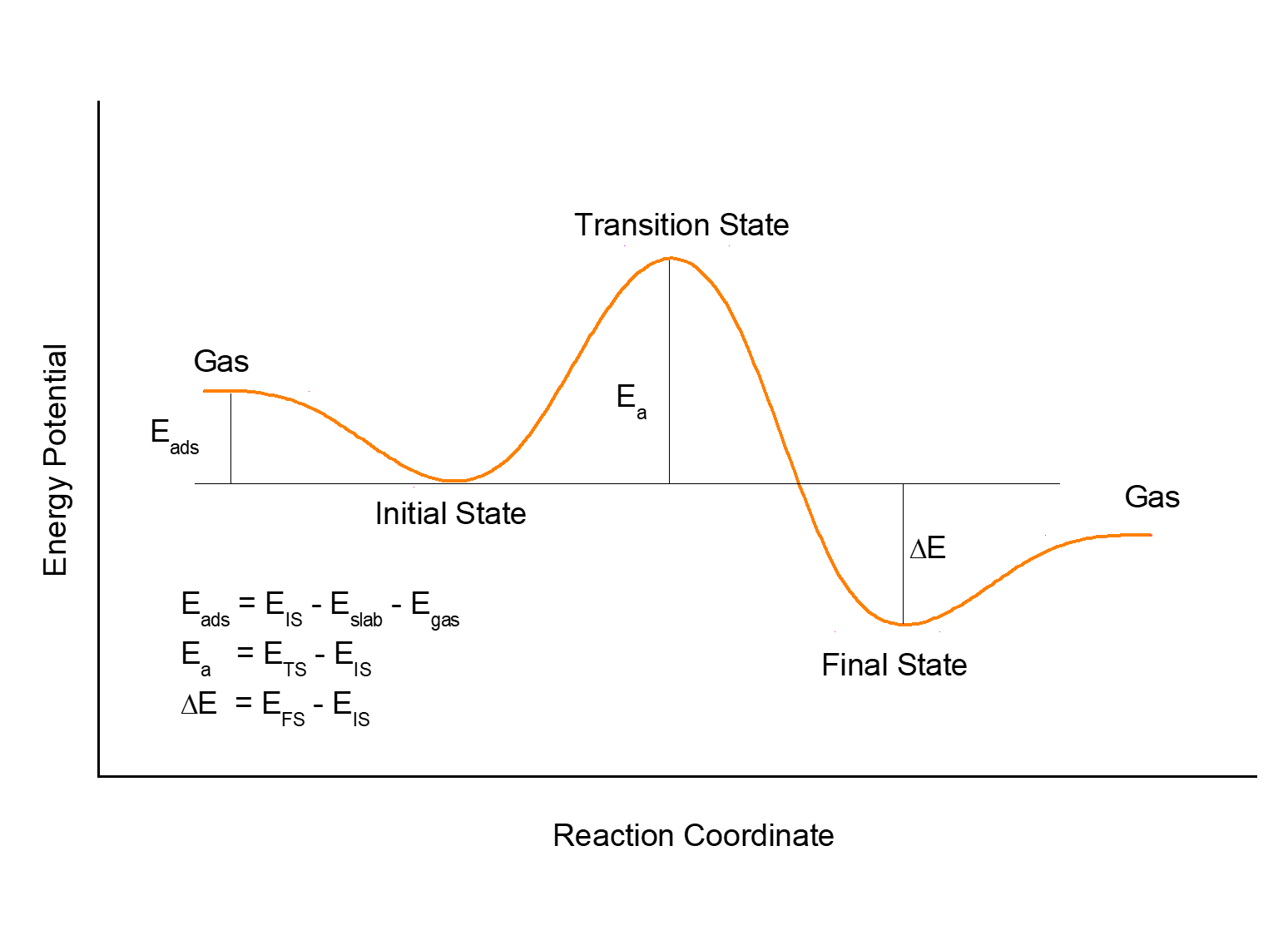
2.2 具体到分子在金属表面上的扩算,一般来说,扩散的能垒差不多是吸附能的12%左右。这里我们说的吸附能,指的是最稳定的那个结构所对应的。
为什么是12% 呢? 这只是一个经验性的结论,大家可以参考大牛Manos Mavrikakis的俺狗娃文章: (不要留言问我要这个文章…)
A Simple Rule of Thumb for Diffusion on Transition-Metal Surfaces
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.200602223

2.3 从结构角度来看,举个面心立方的金属(111)面为例,表面上有fcc, hcp, top, 和 bridge这四个位点。如果fcc或者hcp的位点对应稳定的吸附构型,那么bridge的这个位点就是扩散的过渡态。所以,如果你想简单算一个扩散的过渡态,直接算bridge的吸附就可以了。但是,bridge的位点也不是那么容易就可以算出来的。前面我们讲过Cu(111)表面的吸附计算,很多人就反应bri的结构优化不出来。Bridge 的位点就跟下面的这个独木桥类似,少一不小心就掉下去了。

换个角度来分析:既然我们直接优化得不到bridge的结构,而这个结构恰恰就是过渡态,那么我们就可以使用算过渡态的方法来得到bridge的结构。当然,能直接优化出来bridge的结构是最好的。
过渡态计算的步骤
3.1 第一步:优化初始和末态结构。
首先我们先优化一个扩散前后的两个结构: H在FCC和HCP位点上的吸附。这个对于认真练习过前面计算的筒子们来说就是小菜一碟了。直接列出来top view 的示意图:
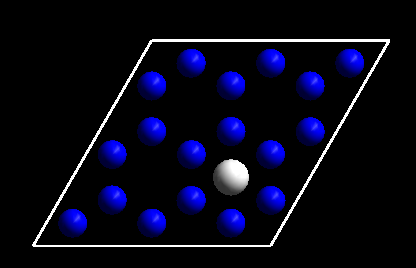
FCC site

HCP site
3.2 第二步:准备NEB的结构
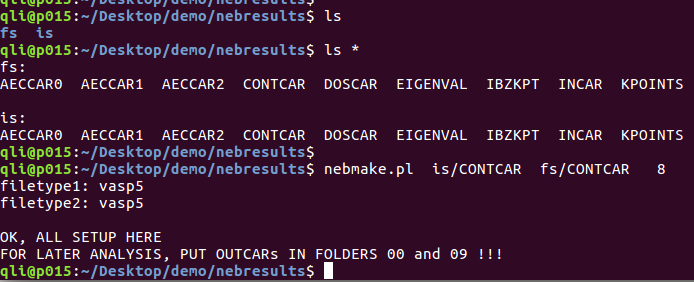
这里我们需要用到VTST官网的一个小脚本:nebmake.pl,使用方法如下:
nebmake.pl IS FS N
1) 敲命令nebmake.pl
2) IS 指的是初始结构:
3) FS指的是末态结构
4) N 指的是你要插点的个数。
细节1:
IS, FS 是VASP的POSCAR或者CONTCAR。
可以是其他目录里面的POSCAR或者CONTCAR, 也可以是当前目录下的POSCAR或者CONTCAR。
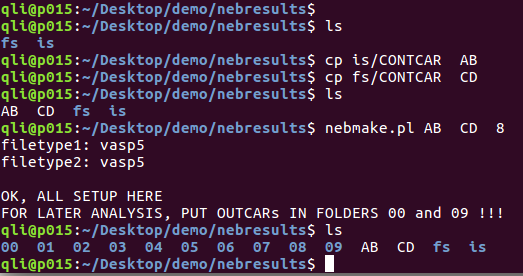
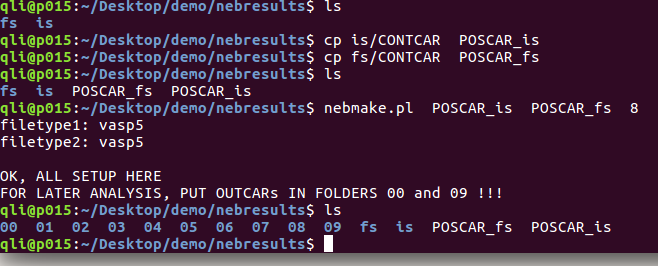
你可以把初始结构的POSCAR命名成你的名字:bigbro , 末态命名成:bigbra. 运行的时候命令应该这么敲:nebmake.pl bigbro bigbra 8
下面图里面的三个事例,是等价的。自己随意领会:
细节2:
插入的点数要保证可以被使用的核数整除。比如我们打算用24个核进行计算,那么N可以是下面的几种情况:
N | Cores used for each Image | Total |
1 | 24 | 24 |
4 | 6 | 24 |
6 | 4 | 24 |
2 | 12 | 24 |
12 | 2 | 24 |
3 | 8 | 24 |
8 | 3 | 24 |
注意:
表中最后一个很可能会出错,因为你用3个核来计算一个image. 上面说的不是绝对的,具体要根据自己的服务器和习惯来设置。
本人使用4个节点(每个节点12个核,共48个核)进行计算,一般N设置位4或者8.
细节三:
同样核数下,N设置的越大,计算每个image所需要的核数就会减少,导致计算变慢。
N需要怎么设置才好呢?
一方面需要大家去咨询一些现成的经验:问问师兄师姐,没有师兄师姐,就在现有的条件下自己去摸索摸索。当然这需要耗费大量的时间和精力,也不符合当下很多人急于速成的心态。
我们着重讲解另一个方面,也就是你对自己研究体系的掌控性。个人感觉分两点:
1)反应过程结构变化的理解,这需要我们在前面初末态的优化上下功夫;
2)化学键的理解:对一个化学键来说,它断裂时候的过渡态键长应该在这个化学键的1-2倍之间。1 指的是这个化学键本身,2指的是这个键被拉长了2倍。你肯定会说我在扯蛋,如果更经验性一点,应该在1.5倍左右。掌握这个这有助于你第一眼去判断自己的过渡态对不对。当然,1.5只是个粗糙的数值,不同的键会有不同的经验性数值。
3.3 第三步: 检查初末态结构的原子坐标是否是一一对应。
这是很多人算过渡态经常忽略的一步,有时候也是很费精力的一步。一个人是不是在闭着眼瞎算,从这一步基本上就可以看出来了。这里大师兄推荐一个linux下检查结构的方法。 命令:p4v 0*/POSCAR 一次性打开所有的Image结构,然后逐个点开,查看整个反应轨迹进行检查。
扩展练习
本节我们需要做的很简单
1) 浏览VTST的网站,阅读相关过渡态计算的步骤以及CI-NEB相关的文献;
2) 下载VTST的脚本,复制到~/bin文件夹,修改nebresults.pl脚本;
3) 计算H在FCC 和HCP的吸附;
4) 使用脚本生成NEB计算的Images文件。
5) 使用脚本生成前面两节:NH3翻转,以及乙烷旋转的Images文件。
总结
本节应该够新手们练习一阵子的了,下一节,我们介绍怎么把NEB的计算运行起来。