Ex62 吸附能的计算(七)
前面我们得到了不同吸附位点上吸附能的顺序,但结构是什么样子的呢?我们优化完的结果对不对?这还是一个问题。因此我们需要查看一下优化完的结果。
1 获取(下载)CONTCAR
没有结构,我们看个屁啊?所以第一步就是把超算中心的计算结果下载到自己的电脑里面。这里我们说获取或者下载CONTCAR,而不是OUTCAR等其他VASP的输入文件,原因在于本人这边网速传输太慢了。所以我的策略是能量等信息在服务器里面直接获取,结构的话只下载CONTCAR。如果网速允许的话,可以把所有的计算结果下载到自己电脑里面,这样查看更加方便。
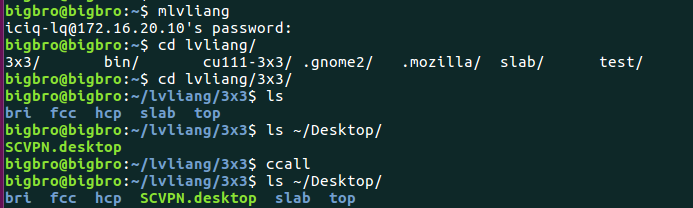
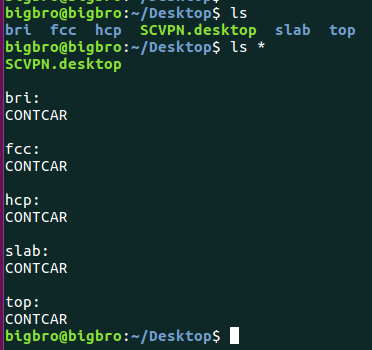
上图中,我们先挂载超算中心到本地电脑上,然后将计算目录下的CONTCAR复制到本地桌面上。(ccall 这个命令)
1 | for i in * ; do if [ -e $i/CONTCAR ]; then mkdir ~/Desktop/$i; cp $i/CONTCAR ~/Desktop/$i ; fi; done |
备注:
由于本人这边传输很慢,即使挂载了超算中心到本地电脑上,访问内容的时候,后台依然有数据传输。所以先下载再查看。
2 使用ASE查看结构:
ASE 是Atomic Simulation Environment的简称,下载安装见:https://wiki.fysik.dtu.dk/ase/ 本人只会在Linux下面使用,Windows用户自行解决。解决不了,我也没有办法。如果Linux用户解决不了,那么使用后面的第二种方法:p4vasp查看结构。
如果你的网速很给力,可以直接通过自己电脑进入超算中心的目录,进行下面的操作。
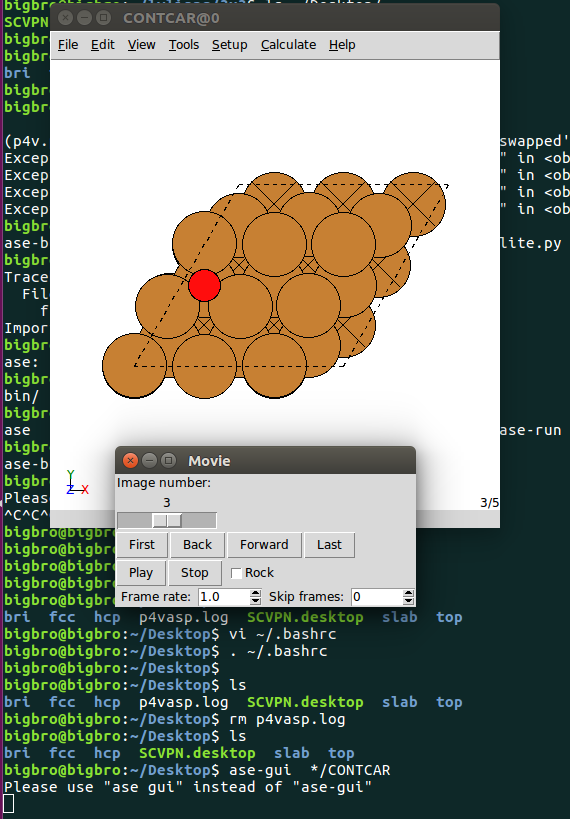
这个软件的优点就是: 我们可以一次性打开当前目录下,所有计算的CONTCAR, 从而避免了使用软件挨个导入结构查看。无形中会减少我们很多的工作量。
1 | ase-gui */CONTCAR |
3 使用p4vasp 查看结构
Ubuntu下面唯一推荐的软件:下面图片拍的不是很好,大家凑活着看吧。
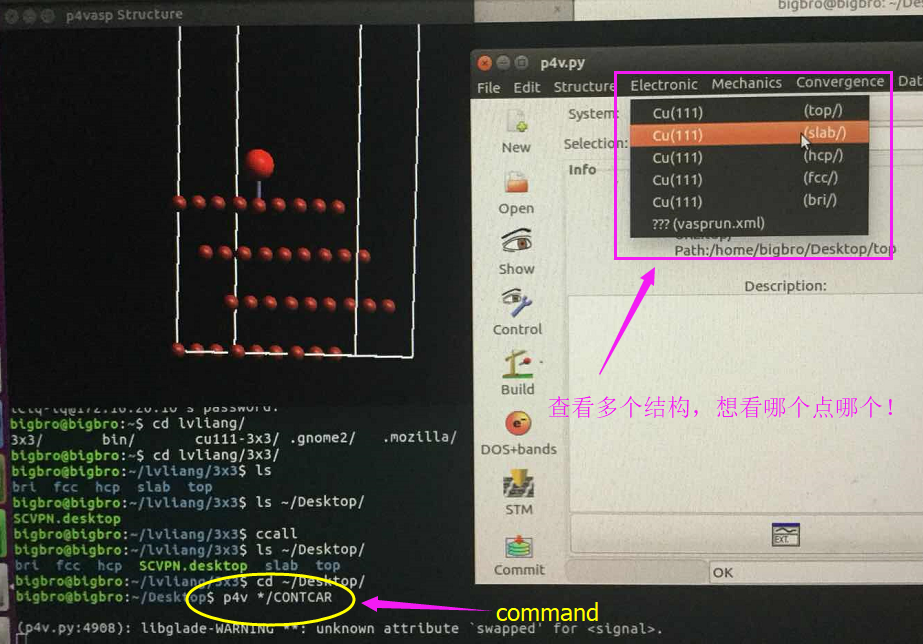
这里,p4vasp和前面说的ASE一样,也可以一个命令打开所有的计算结果。
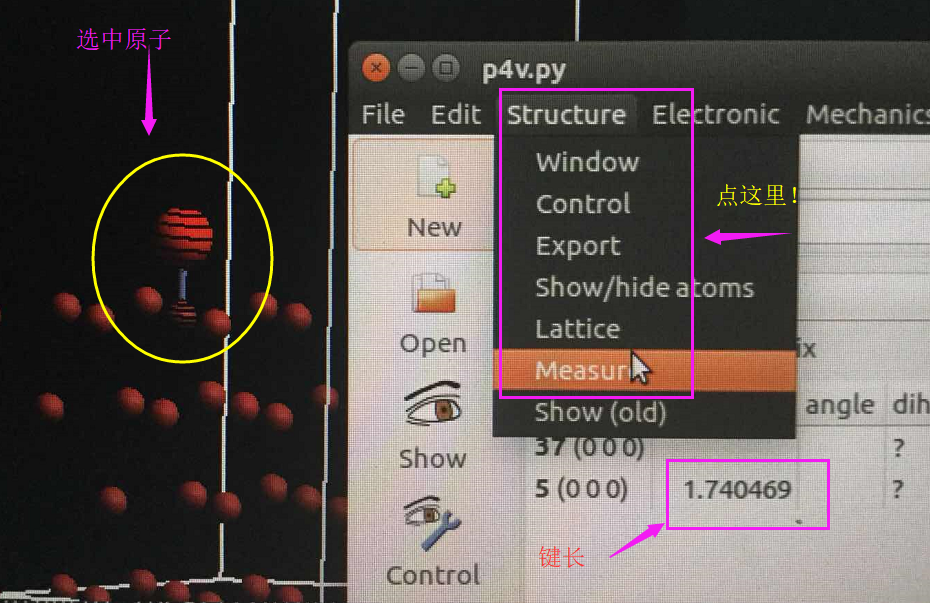
作为p4vasp的忠实粉丝,这也是本人唯一推荐的Ubuntu系统下查看,搭建结构的软件。
- i)使用p4vasp可以非常容易地进行原子替换,平移,旋转等基本操作。
- ii)可以查看VASP的结算结果,DOS,能带,优化过程等等。
- iii)Windows 系统下p4vasp的功能有些弱,除了不能批量打开文件外(可能是本人不会用),其他的和Ubuntu差不多。
- iv)这个软件也有很多其他细微不尽人意的地方,但不影响我们的正常使用。
- 如果你刚开始接触这个软件,认真用鼠标各个地方点点操作一下,查看各个功能按钮的作用。
- 此外,VASP官网的
ppt教程中也有一些零星的p4vasp操作教程,大家可以参考一下。
4 其他软件:
当然了, 不论在linux还是Windows下面,都有很多查看结构的软件,比如:Jmol,Xcrysden,Molden, VESTA, Material Studio等等。这里就不再详细介绍了,主要原因是本人不太会使用这些软件操作。目前大家需要做的就是根据自己的喜好,掌握一个软件:学会查看结构,键长,键角等信息即可。切记不可贪多,等一个软件掌握好了之后,有余力的话再去学习另一个的操作。
5 扩展练习:
1) 自己优化O在Cu(111)表面上不同位点的吸附,计算吸附能
2) 选择一款自己喜欢的软件,查看不同的吸附结构。
3) 思考其他单原子在其他金属表面上的吸附,该如何计算?
4) 思考原子在表面上,为什么不同吸附位点的吸附能不一样?
6 总结:
本节没有什么技术难度,全靠自己亲自手动操作,使用一个软件并不是一蹴而就的过程,大家先把基本的简单操作掌握了,后面再逐渐提高自己的其他技能。此外,本节学习完之后,单原子在表面上的吸附对大家来说应该不是什么困难的事情了。