在ex13中,当初始值为1.5 $\AA$ 的时候,计算共进行了9步。对比下之前我们采用实验值(Ex11)作为初始结构计算时用了4步。从这里我们可以看出来,如果你一个合理的初始结构,可以加快优化的速度,减少机时,节约你的时间。当然,计算的具体时间可以通过OUTCAR尾部的信息查看。
不同初始结构对结果的影响。 那么不同的初始结构,除了在时间上,对计算的结构还有影响吗? 首先我们对比下Ex11和Ex13的结果。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW$ tail ex13/nsw10/CONTCAR 0.0000000000000000 8.0000000000000000 0.0000000000000000 0.0000000000000000 0.0000000000000000 8.9000000000000004 O 2 Direct 0.0000000000000000 0.0000000000000000 0.0149145061380336 0.0000000000000000 0.0000000000000000 0.1536248197046605 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW$ tail ex11/opt/CONTCAR 0.0000000000000000 8.0000000000000000 0.0000000000000000 0.0000000000000000 0.0000000000000000 8.9000000000000004 O 2 Direct 0.0000000000000000 0.0000000000000000 -0.0019537557431563 0.0000000000000000 0.0000000000000000 0.1367852164173130 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW$
在Ex11和Ex13中,两个计算的能量和CONTCAR中的键长值也几乎相等,看下面的结果。
1 2 3 4 5 6 7 8 9 10 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW$ python Python 2.6.6 (r266:84292, Sep 4 2013, 07:46:00) [GCC 4.4.7 20120313 (Red Hat 4.4.7-3)] on linux2 Type "help", "copyright", "credits" or "license" for more information. >>> 0.1536248197046605-0.0149145061380336 0.13871031356662689 >>> 0.1367852164173130--0.0019537557431563 0.13873897216046929 >>>
另一个bad结构的计算 上次我们用了一个大于实验值的初始结构1.5$\AA$。下面我们看下小于实验值的初始情况:0.9 $\AA$ 。
1 2 3 4 5 6 7 8 9 10 11 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/ex14$ cat POSCAR O 1.0 7.5 0.0 0.0 0.0 8.0 0.0 0.0 0.0 8.9 O 2 Cartesian 0.0 0.0 0.0 0.0 0.0 0.9
INCAR,KPOINTS,POTCAR等不变,提交任务,等待计算结束,查看结果。
1 2 3 4 5 6 7 8 9 10 11 12 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/ex14$ tail OSZICAR DAV: 18 0.141396114006E+03 0.20879E-02 -0.88760E-04 144 0.130E-01 0.579E-01 DAV: 19 0.141397297948E+03 0.11839E-02 -0.52751E-04 96 0.893E-02 0.455E-01 DAV: 20 0.141388687914E+03 -0.86100E-02 -0.28657E-03 144 0.231E-01 0.206E+00 DAV: 21 0.141395321236E+03 0.66333E-02 -0.62073E-03 96 0.361E-01 0.112E+00 DAV: 22 0.141389669824E+03 -0.56514E-02 -0.95109E-03 96 0.429E-01 0.161E-01 DAV: 23 0.141382943017E+03 -0.67268E-02 -0.22808E-03 96 0.216E-01 0.118E+00 DAV: 24 0.141390086243E+03 0.71432E-02 -0.39635E-03 96 0.307E-01 0.825E-01 DAV: 25 0.141385767811E+03 -0.43184E-02 -0.44984E-03 96 0.305E-01 0.480E-01 DAV: 26 0.141385726226E+03 -0.41585E-04 -0.10329E-04 96 0.429E-02 5 F= 0.14138573E+03 E0= 0.14138680E+03 d E =0.141123E+03 mag= -2.0000
计算共进行了5步,且最后的磁矩看起来是正确的。思考 : 从这里得出的信息,能确定我们的计算结果是正确的吗?
答: 不知道。因为我们还要要去查看一下结构。判断结构是否合理。如果结构不合理,则收敛的计算也是失败了。
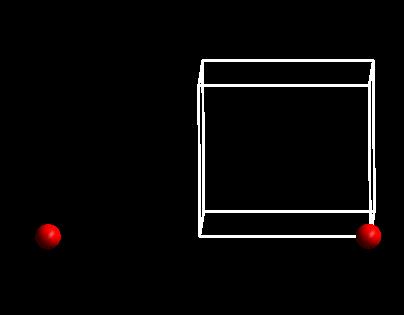
用p4vasp打开CONTCAR后,如下图:
师兄,这是神马情况,两个原子怎么跑这么远?
不用担心,这是因为周期性的原因。p4vasp中可以进行如下的操作:
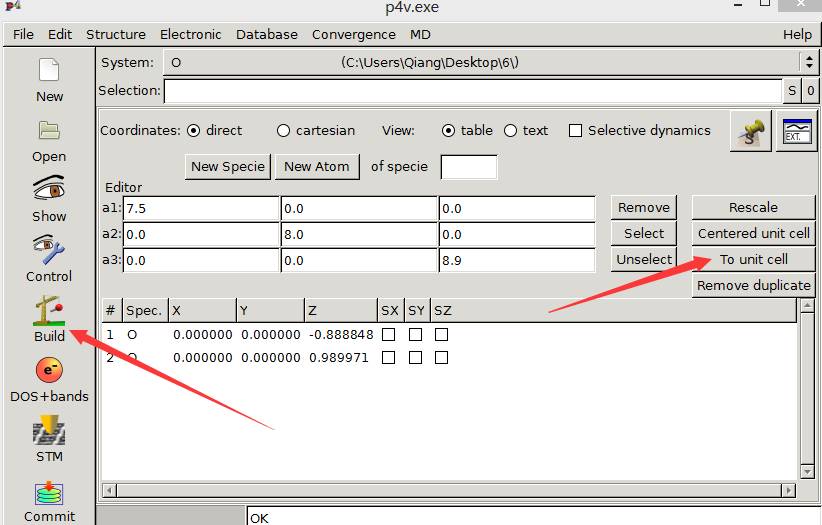
首先,点击左侧的Build按钮,然后再点击右侧的 To unit cell。这样你会发现结构调整到下图的样子:
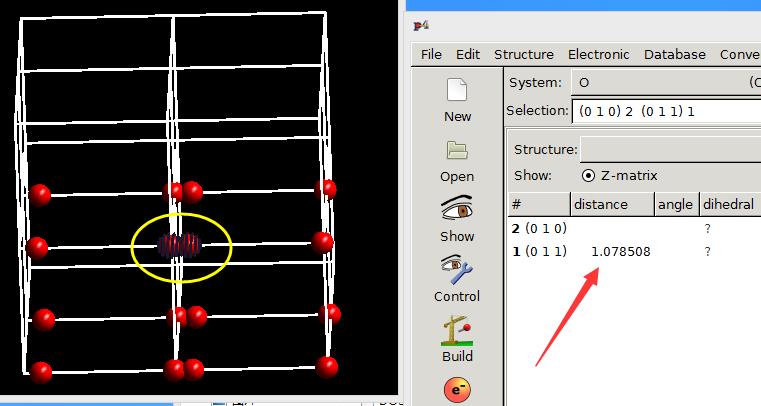
两个原子之间的距离还是很长(7.821 $\AA$),但实际键长不是这么长的。
而是8.9-7.821 = 1.079 $\AA$。
师兄你为什么这么算?
因为我们的体系是周期性的,也就是图中的格子在三维方向上可以无限重复,如果我们向左重复一个单元,那么在新的单元中右侧的氧原子与原来左侧的氧原子距离很短。已知格子在z方向的长度为8.9 $\AA$,减去7.821就是剩下的两个氧原子之间的键长了。
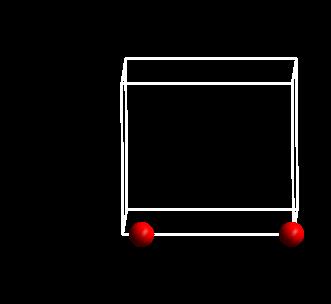
如果,你还不明白,进行下图的操作:
点击左侧的Control 选项,然后在下面红色框中,将格子在三维方向上重复,效果如下:
注意,该操作只是展示三维方向的结构,如果此时你保存结构,不管你在三维方向上重复了多少次,保存的结构则还是原来的尺寸大小。
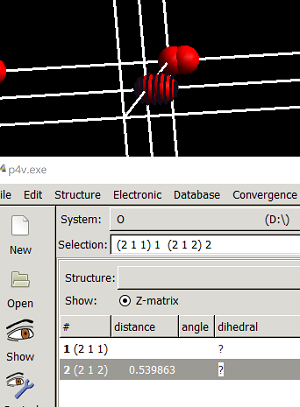
键长为1.0785 $\AA$。
周期性的显示问题 暂且抛开对错不说,由于周期性导致的原子不在一个格子里面的情况,在今后的计算中你会经常碰到。如果你遇到这种情况,不要立即在群里问:师兄,为什么优化之后,体系中的原子不见了?为什么之前左面原子不见了,右侧本来没有原子,优化完多了?
归根结底都是周期性导致的显示问题。你需要做的就是把结构在三维方向上重复一下,查看结构是对还是错。
结果对还是错? 现在我们分析下对错,已知O$_2$分子的键长为1.2075 $\AA$,因此该计算与实验值偏差为:(1.0785-1.2075)/1.2075 =10.68 %,这么大的偏差,是不可以忍受的。
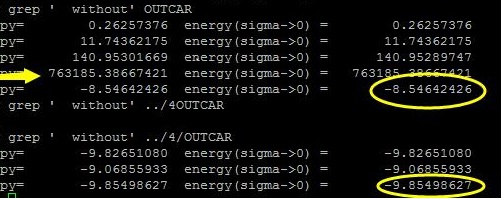
检查一下能量: 为 -8.54642426 eV。 之前正确的能量为: -9.85498627eV。
在第二版的改进中,大师兄又计算了这个任务一次,得到了另外一个结果:2个O原子距离很短。
查看能量:
1 2 3 4 5 6 7 8 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/ex14$ grep ' without' OUTCAR energy without entropy= 0.26254047 energy(sigma->0) = 0.26254047 energy without entropy= 11.76187113 energy(sigma->0) = 11.76187113 energy without entropy= 142.97353506 energy(sigma->0) = 142.97353506 energy without entropy= 1359169.21650280 energy(sigma->0) = 1359169.21650280 energy without entropy= 141.38788142 energy(sigma->0) = 141.38680382 iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/ex14$
思考: 能量为什么会差这么多呢(1.11 eV 或者最新结果140多eV)?
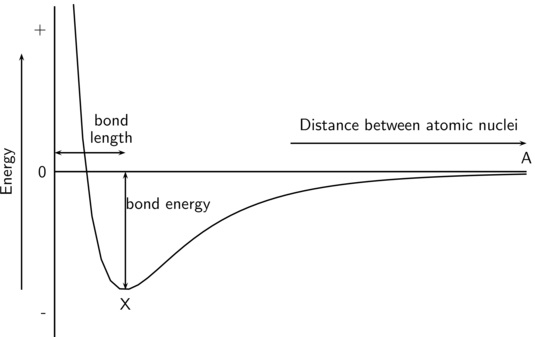
答: 我们需要知道体系的能量随键长的变化关系:如下图:
图中X 处是O$_2$的稳定结构,两个原子间距离小于X处的键长时,它们之间的排斥力导致了体系的能量快速升高。
由于我们已经知道了正确的计算结果,通过分析后,这次的计算失败!但对于不知道结果的时候,怎么判断计算是否成功失败呢?首先根据VASP计算的收敛情况,也就是计算至少应该正常结束,其次,这是远远不够的,我们还要查看输出结构的几何构型,判断是否具有物理或者化学的意义,还要看每一步收敛的能量信息。这些就需要我们化学基础知识了。
扩展练习及思考 1 计算为什么会失败?
2 分析该计算中每一步收敛的情况,以及能量的变化。
总结: 1 不合理的结构会增加计算时间;
2 不合理的结构会导致计算结果没意义;
3 知道怎么处理周期性结构中,原子不在一个晶格里面的情况;
4 学会判断计算结果的物理或者化学意义。