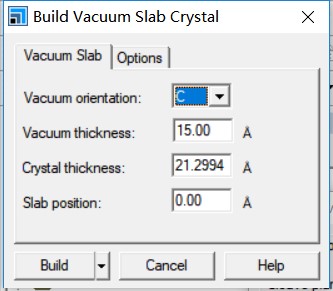
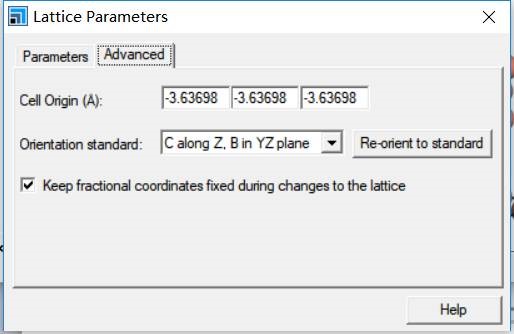
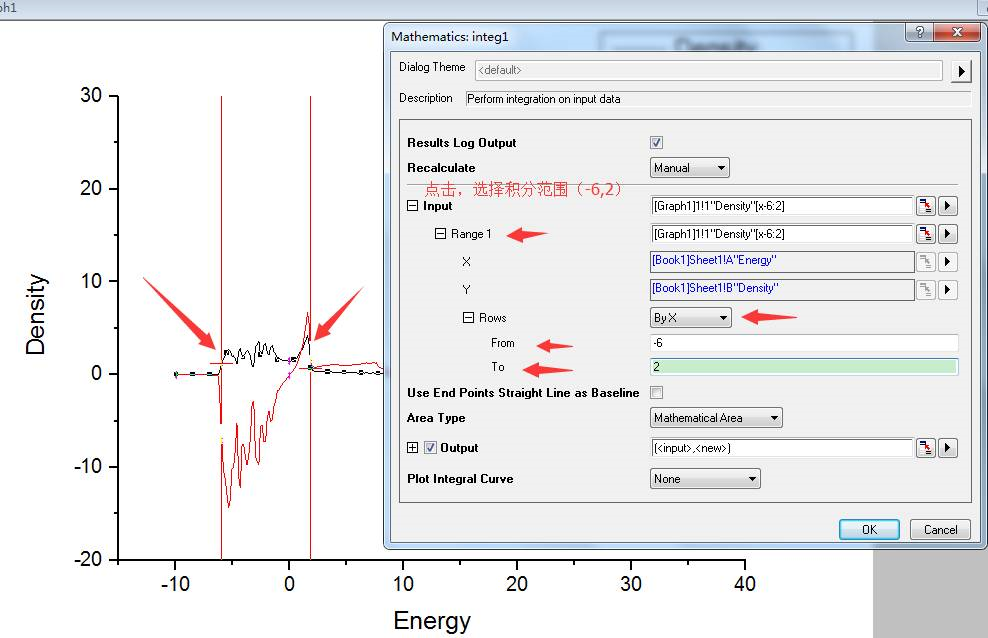
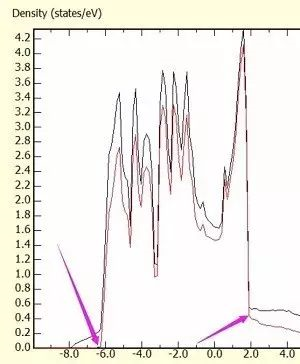
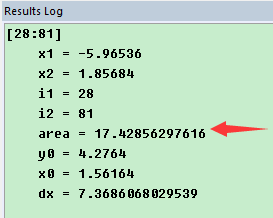
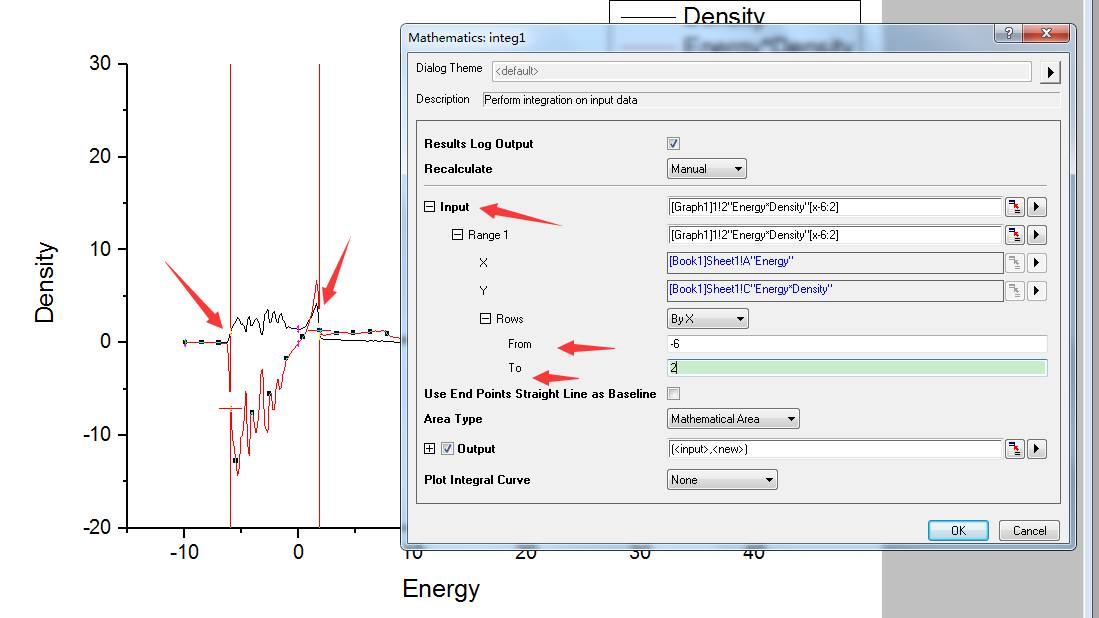
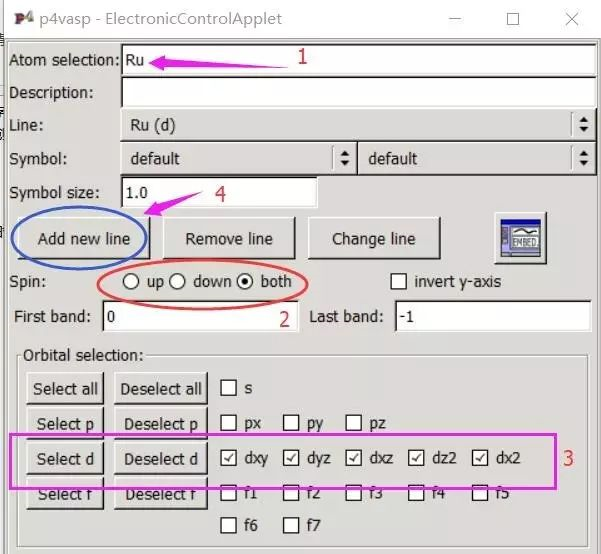
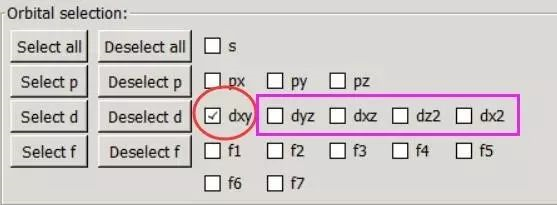
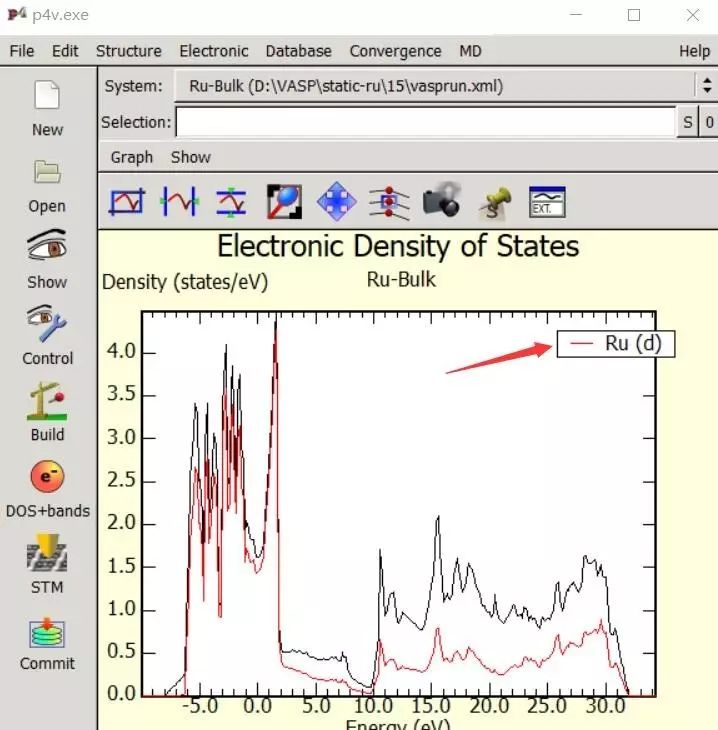
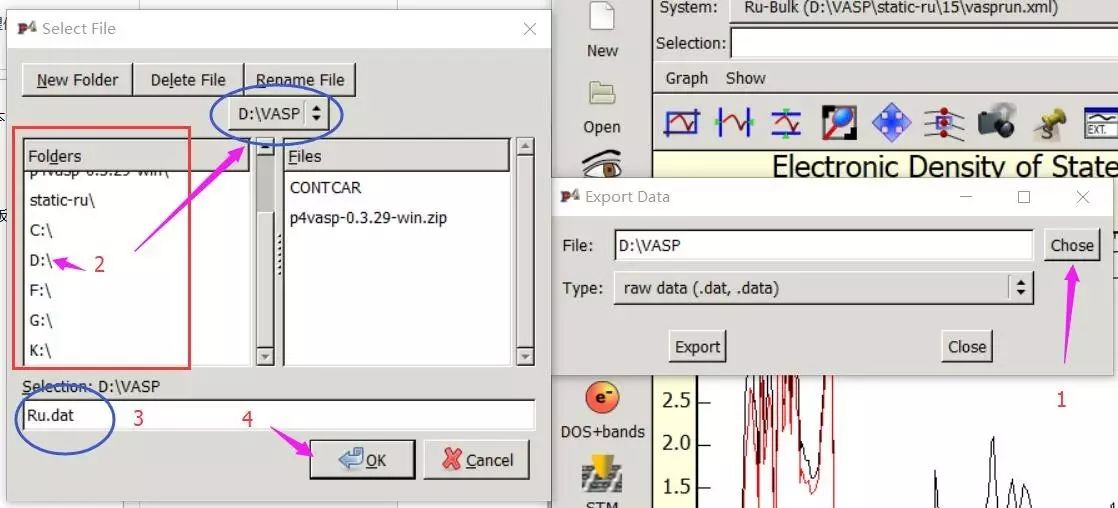
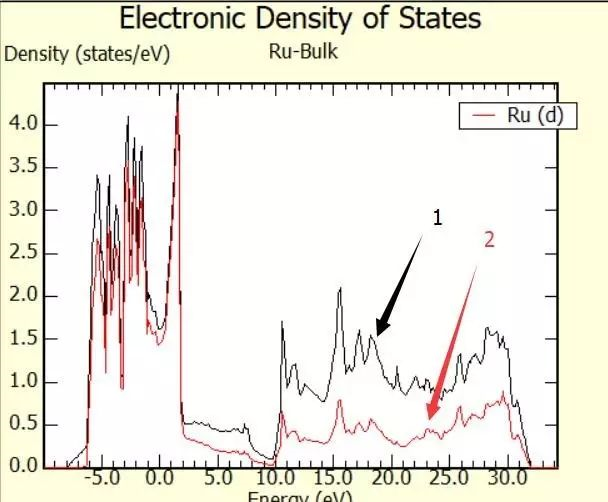
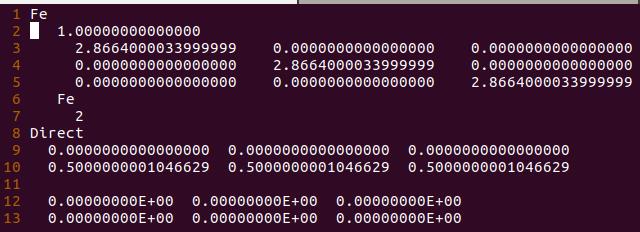
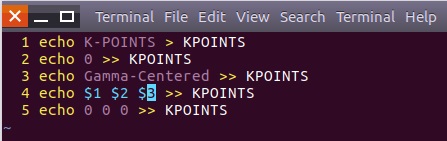
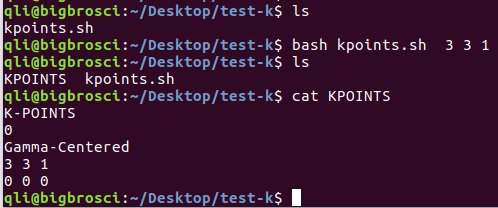
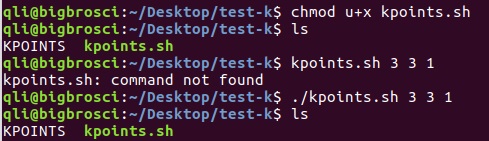
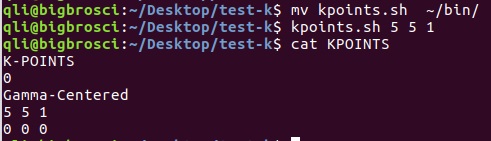
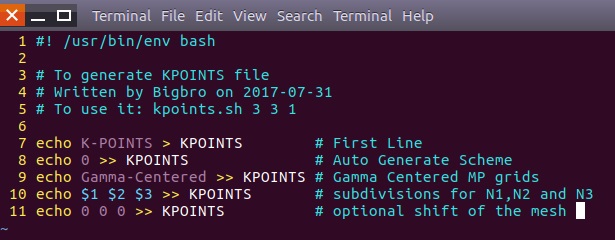
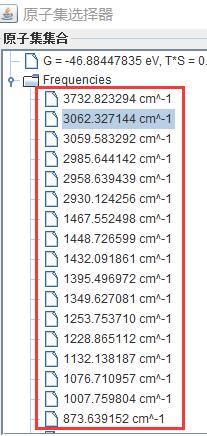
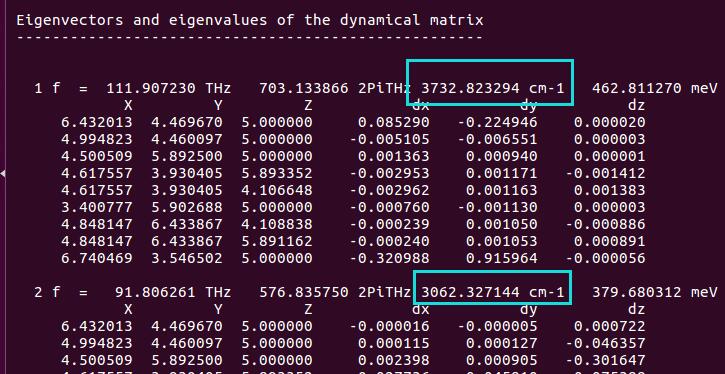
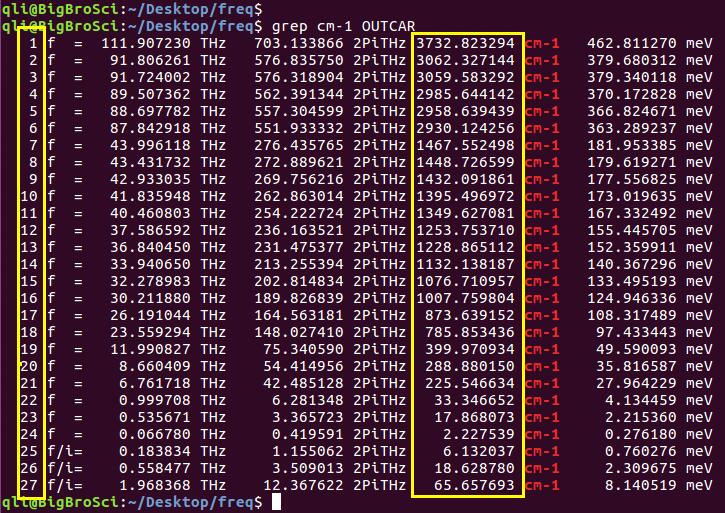
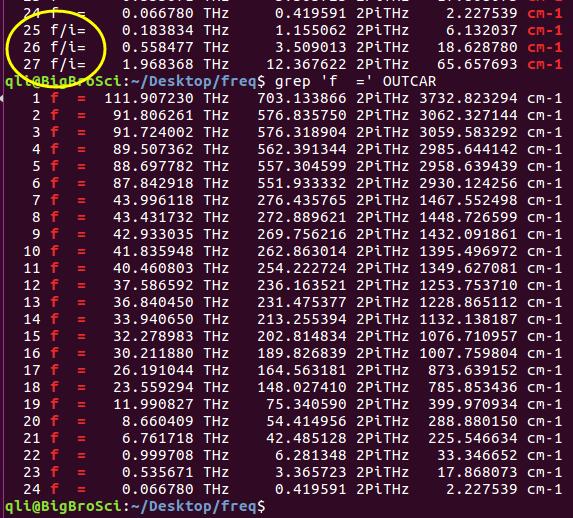
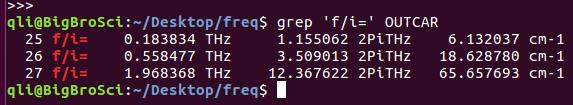
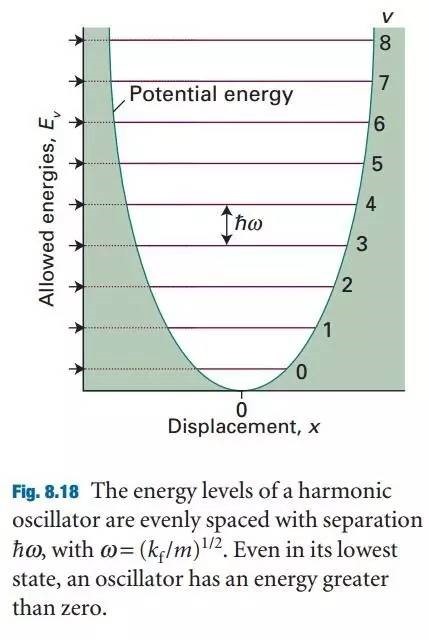
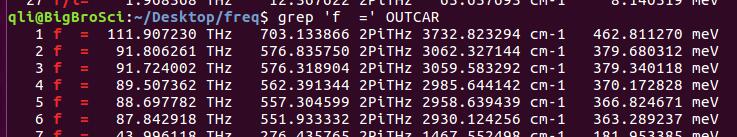
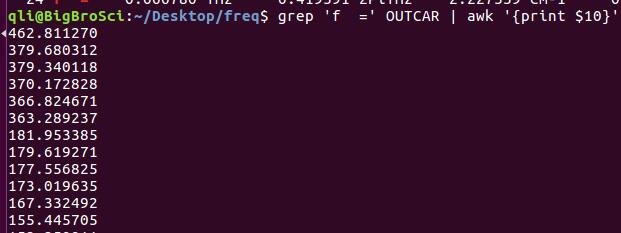
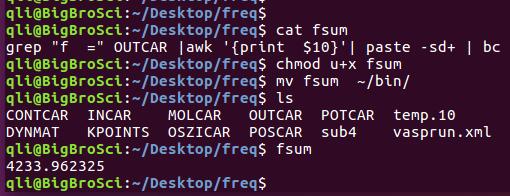
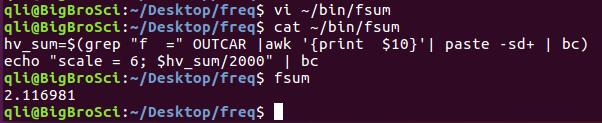
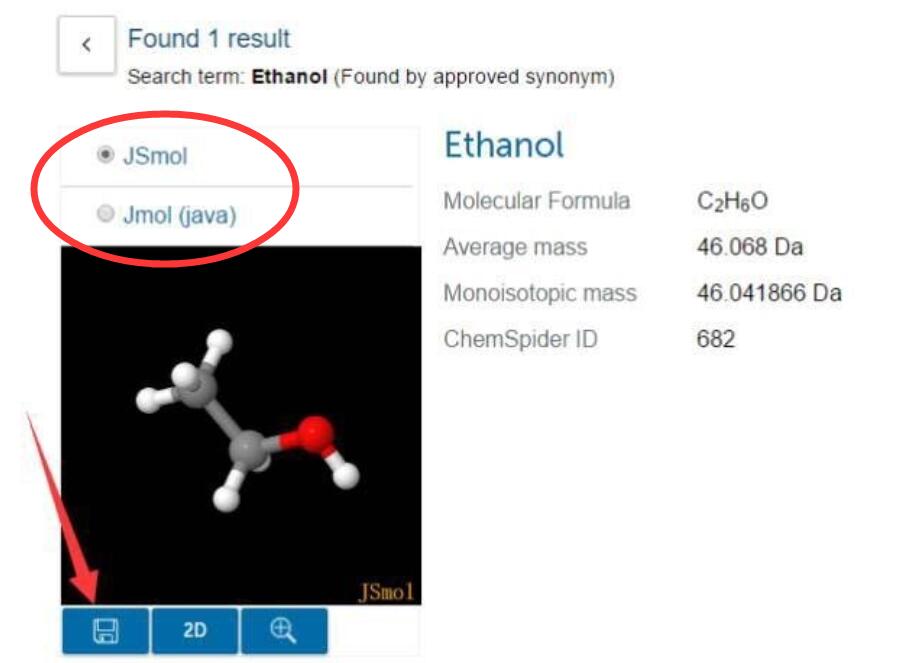
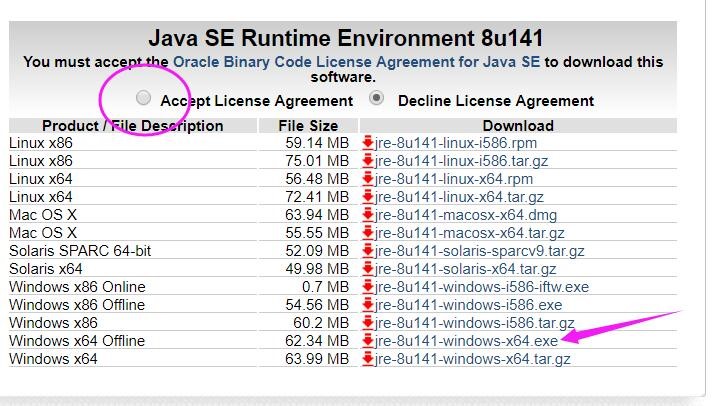
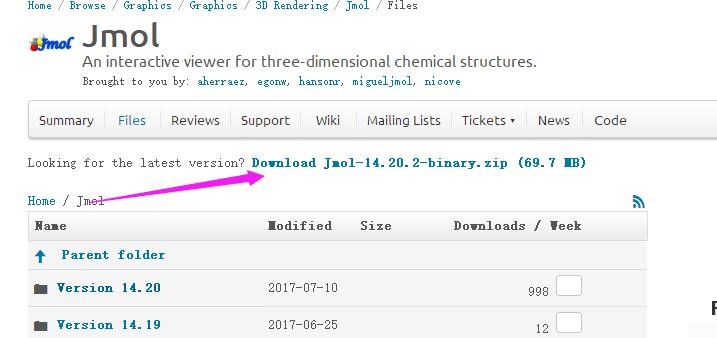
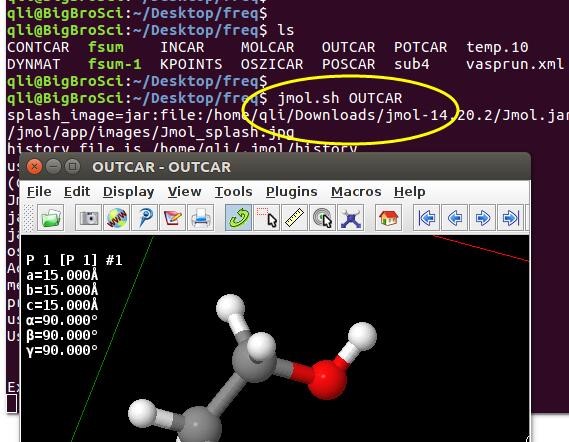
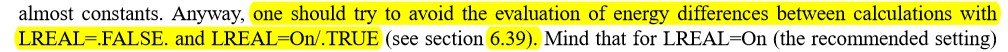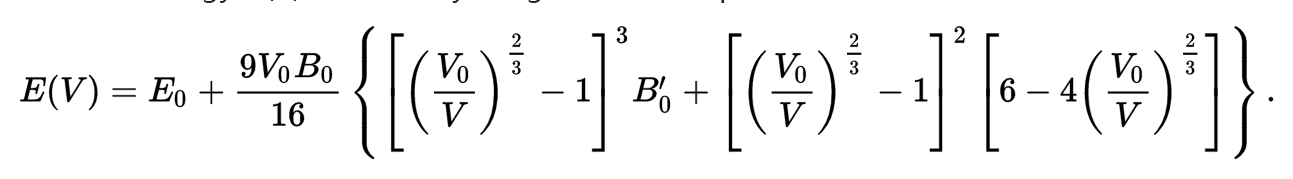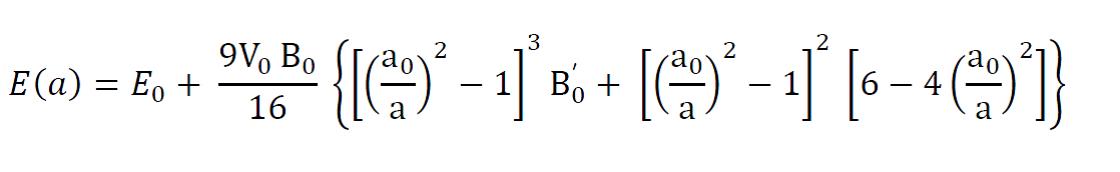为什么在z方向上只用1个K点?查找参考书(DFT:A Practical Introduction, Page 88)的说明: If the vacuum region is large enough, the electron density tails off to zero a short distance from the edge of the slab. It turns out that this means that accurate results are possible using just one k point in the b3 direction.
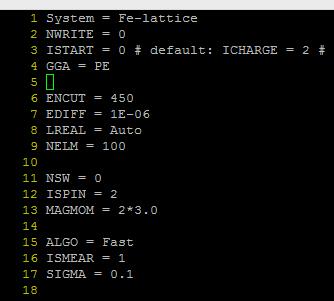
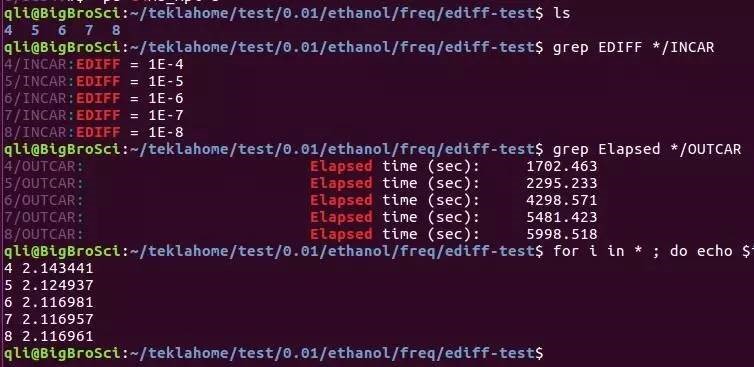
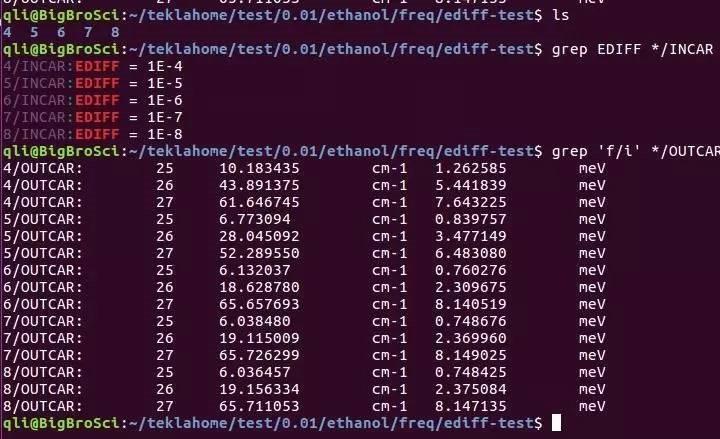
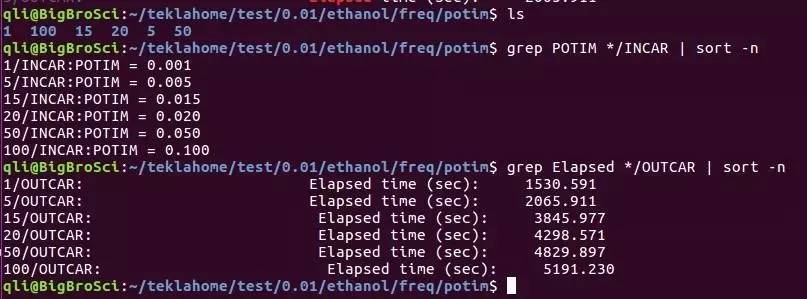
E)这里LREAL设置为Auto,是为了和后面表面能计算统一。计算时要根据原子数的多少,以及后续的计算选择LREAL的值。不能拿LREAL=.FALSE.和LREAL=ON/.TRUE.计算的结果进行能量比较。看下面 F) 注意:块体材料计算时,为了后续计算其他性质,常常要求整个计算中使用相同的ENCUT,ENAUG, PREC, LREAL, ROPT,这部分大家可以看看手册 8.3 What kind of “technical” errors do exist, overview
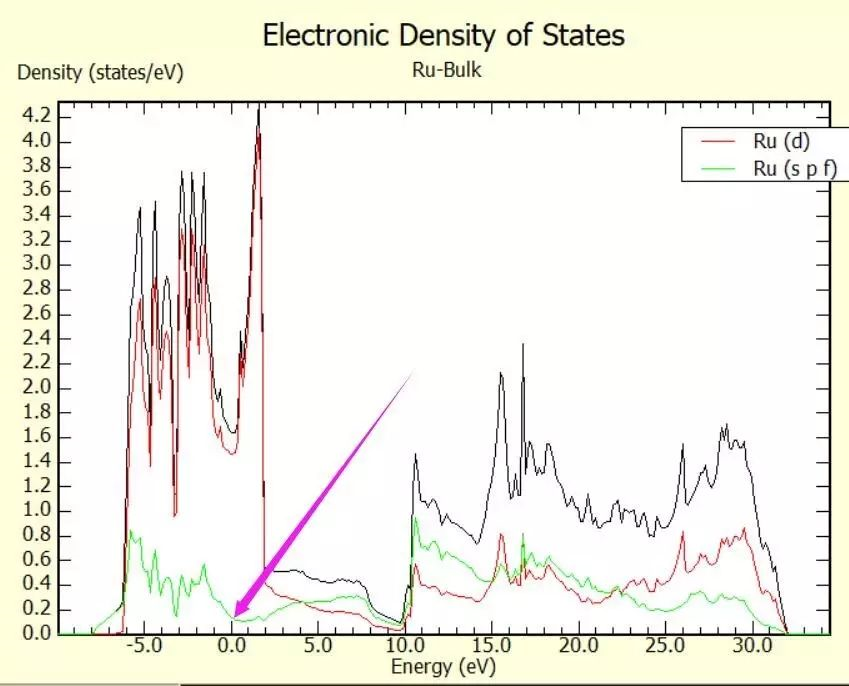
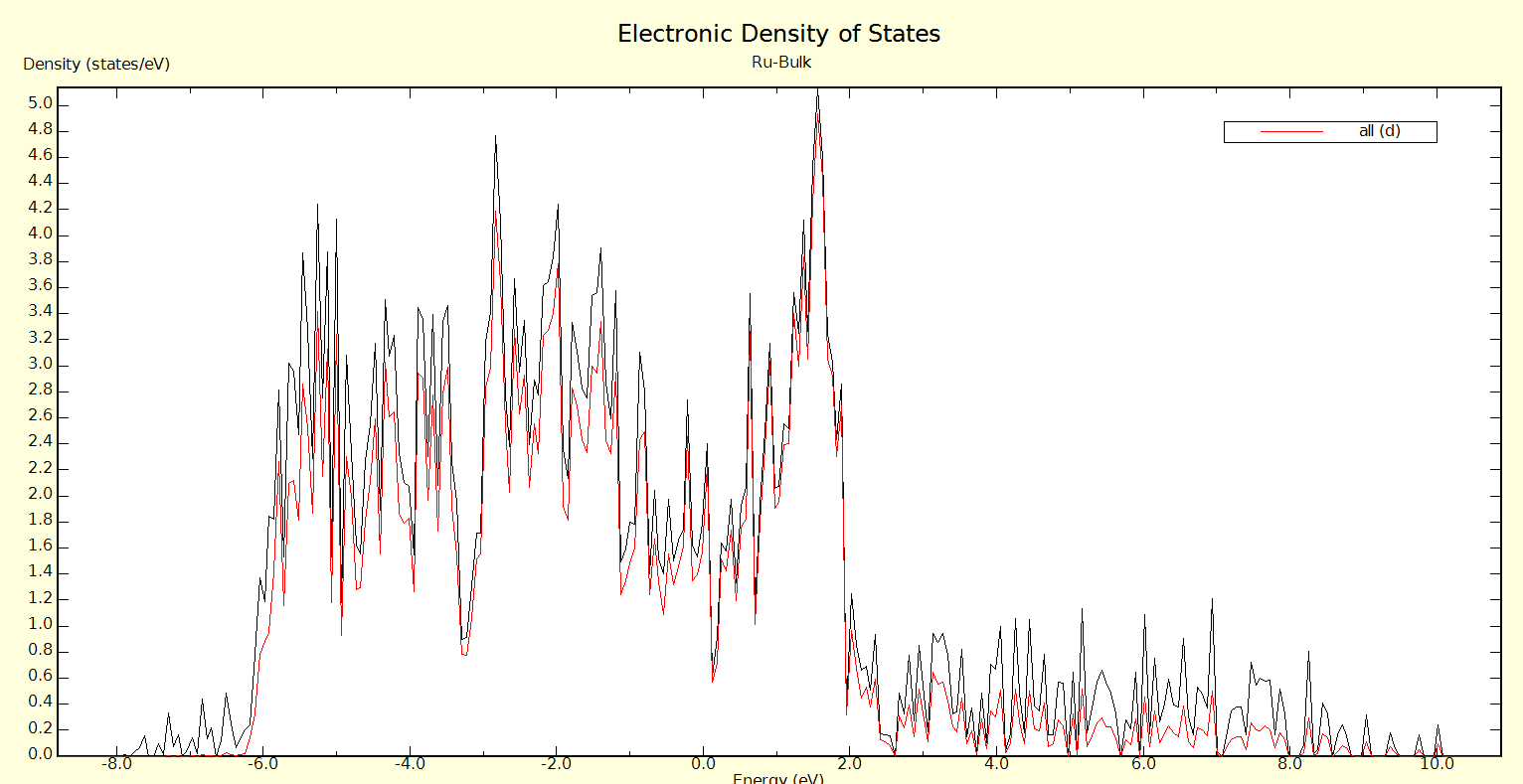
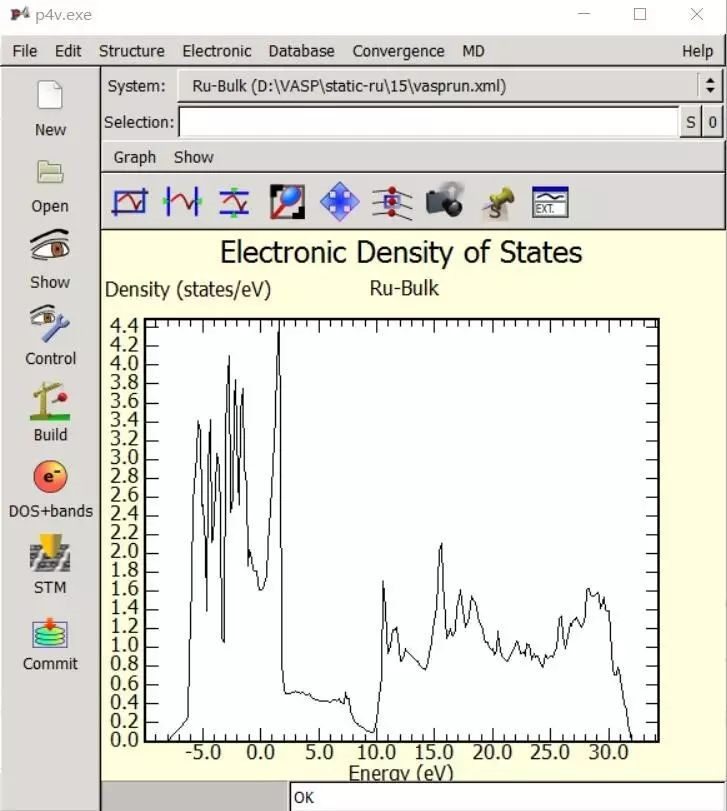
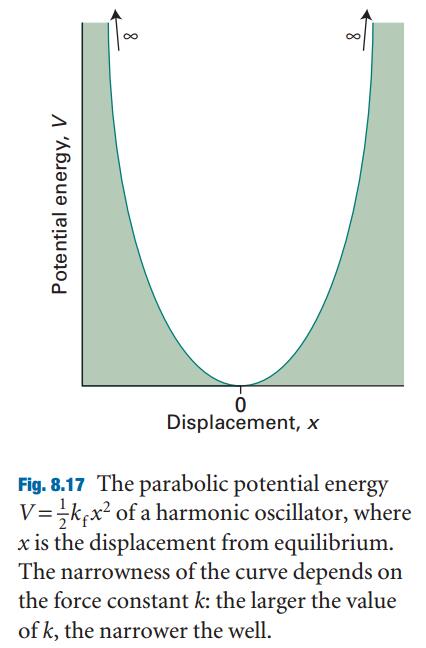
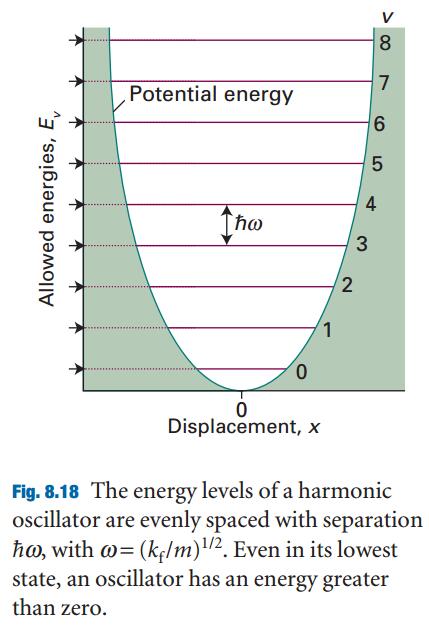
For the calculation of the total energy in bulk materials we recommend thetetrahedron method with Blöchl corrections (ISMEAR=-5). This method also givesa smooth nice electronic density of states (DOS).
For further considerations on the choice for the smearing method see sections 9.4, 10.6. To summarize, use the following guidelines:
For semiconductors or insulators use always tetrahedron method (ISMEAR=-5), if the cell is too large (or if you use only 1 or two k-points) use ISMEAR=0.
For relaxations in metals always use ISMEAR=1 and an appropriated SIGMA value (the entropy term should less than 1 meV per atom). Mind:Avoid to use ISMEAR>0 for semiconductors and insulators, it might result in problems.
For metals a sensible value is usually SIGMA= 0.2 (that’s the value we use for most transition metal surfaces).
For the DOS and very accurate total energy calculations (no relaxation in metals) use the tetrahedron method (ISMEAR=-5).
A plane wave basis set is created for thehexagonal lattice (left), using the reciprocal lattice vectors inside the redcircle. Then the lattice relaxes into a cubic symmetry (right). Keeping the redcircle basis constant results in lattice vectors taken from an ellipsoidinstead of a spherical area (compare to the blue circle).
深层次的俺也不懂,但是PulayStess 有2个办法可以减小:
A) 体积不变,采用一系列的晶格常数计算,然后拟合得到准确的晶格常数;(你已经知道怎么去做了,这么做是为了避免计算时晶格体积发生变化!);
B) 既然是因为ENCUT不够导致的,那么我就使劲增加ENCUT来消除(上图中unless后面的那句)。借用维基百科的一句话:Pulay stress can be reduced by increasing the energy cutoff. 所以,当有人用ISIF= 3计算,而不去设置ENCUT的时候,尽情地去嘲笑他傻X吧!!!感觉不过瘾,还可以继续吓唬他说后面的计算也有问题。
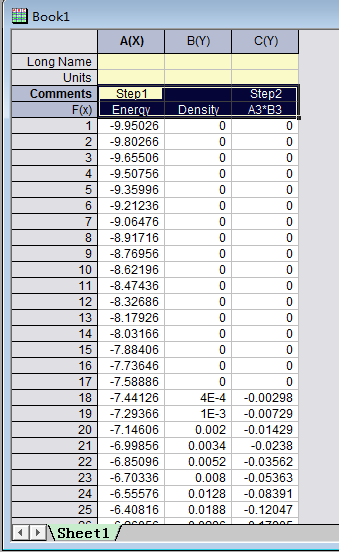
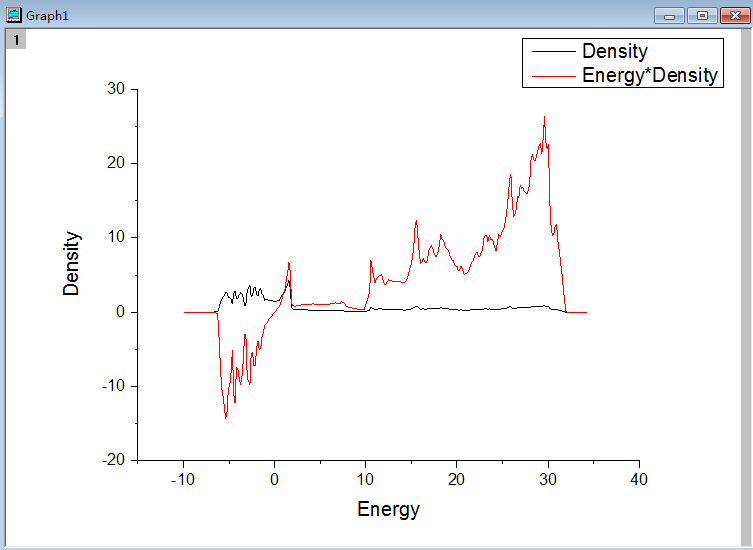
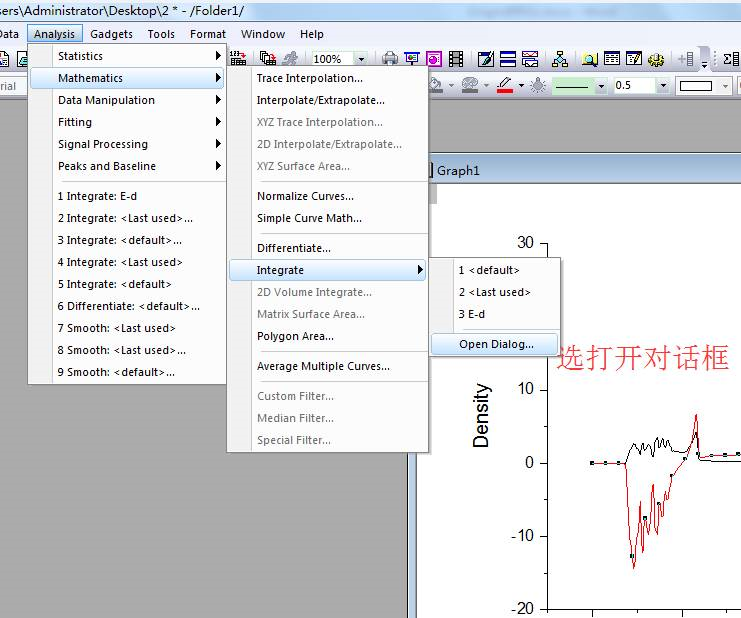
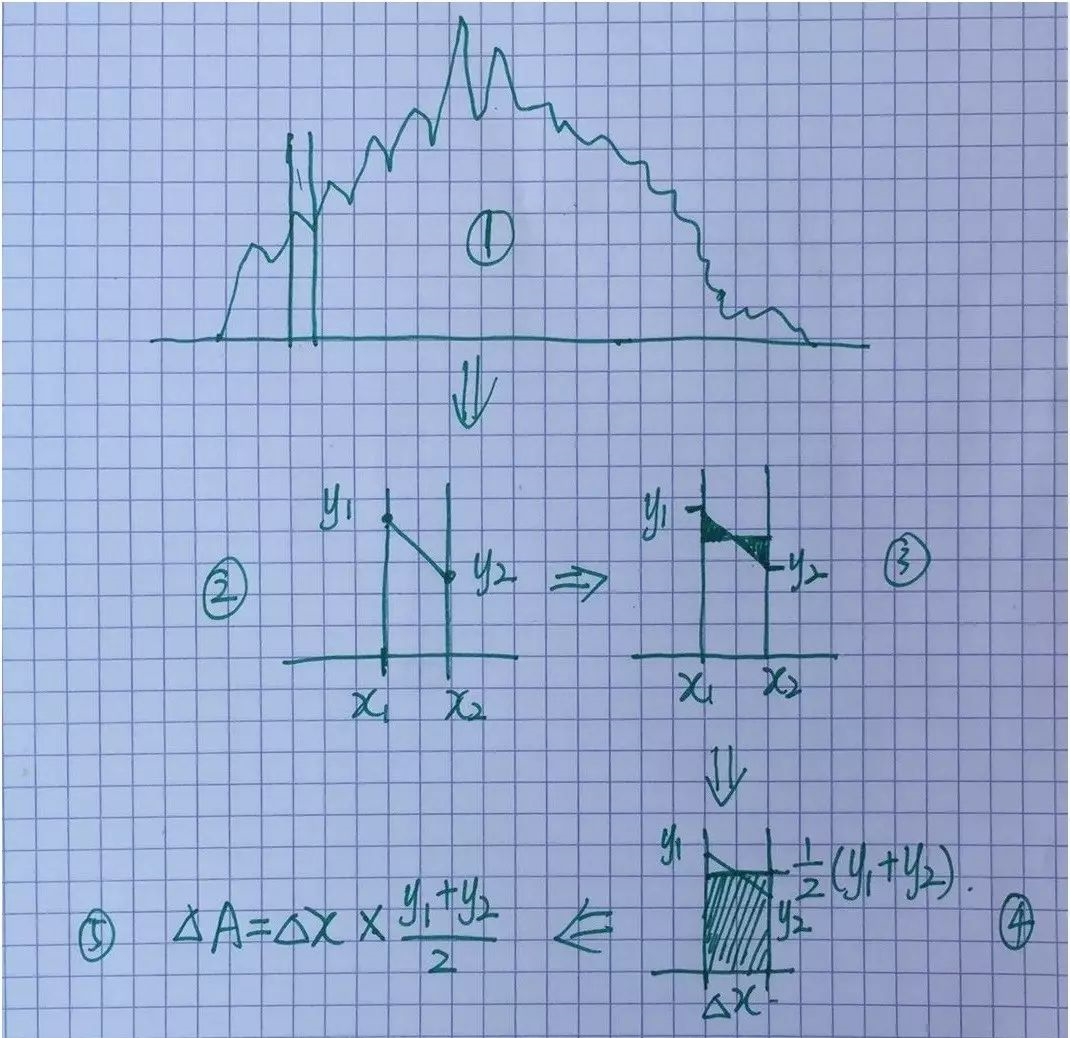
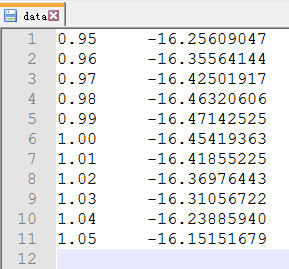
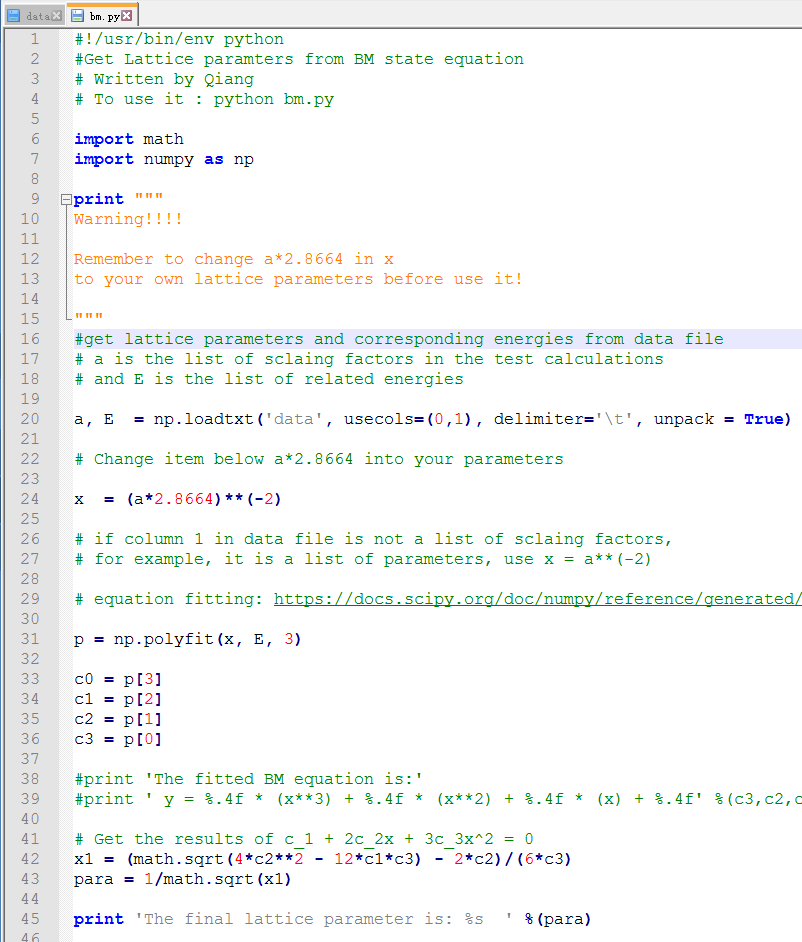
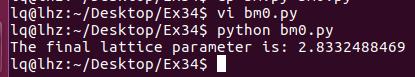
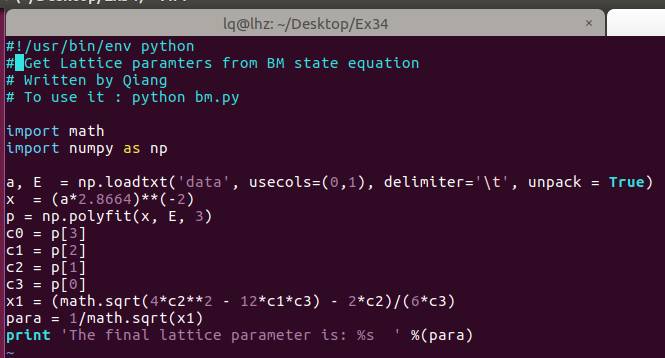
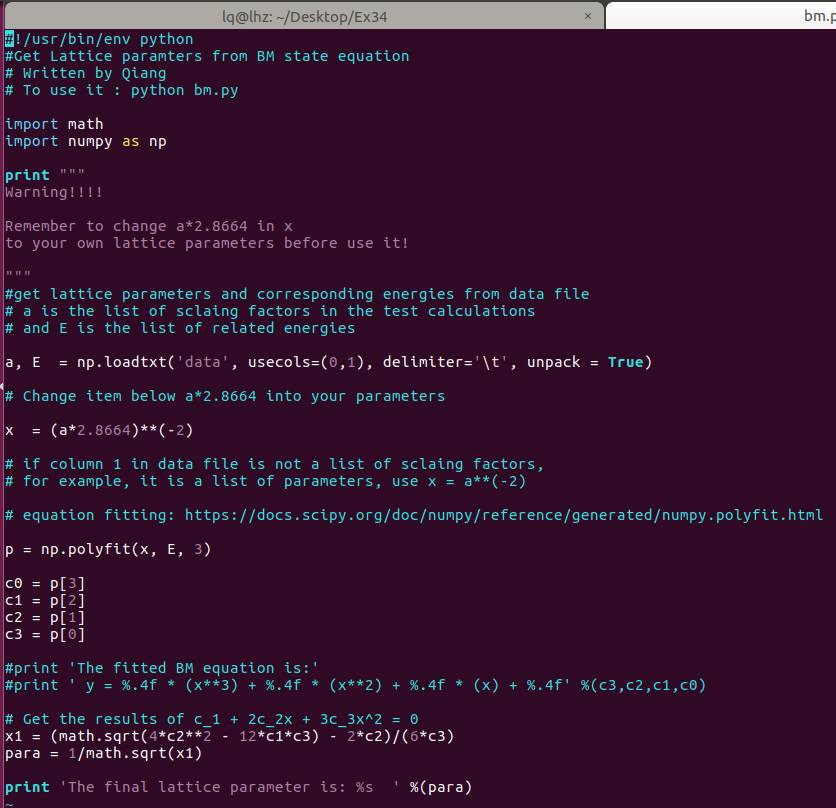
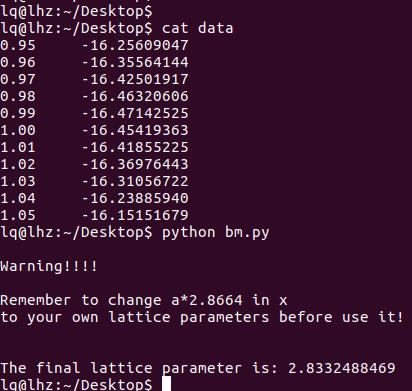
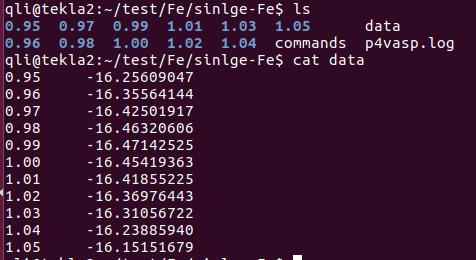
通过第20行,我们可以获得两个数列,第一个是 a 包含了data文件中的缩放系数, 第二个是E,包含了data文件中的能量信息
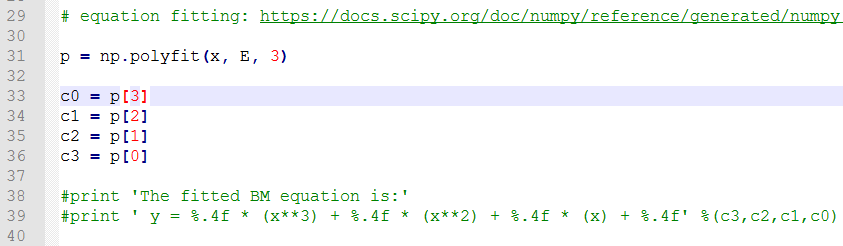
C) 第22-28行,把缩放系数转换为晶格常数(a2.8664),然后平方取倒数(*(-2)),用来得到我们前面讲解的x。这是因为:当我们把BM方程写成: $y(x) = c_0 + c_1x + c_2x^2+c_3x^3 $ 的形式时,前提是令 (1/a)$^2$ = x。不懂的具体见Ex33中的内容。截止到现在,我们有了三个数列: a, E 和 x
最后,新手的话,请认真学习Learn Vasp The Hard Way 这本书(www.bigbrosci.com) 按照大师兄写的内容,从头认真学习VASP(本书只作为参考,关键是去浏览官网!!!);如果在学习中遇到问题,也可以把问题准备好,发送邮件给大师兄解决:lqcata@gmaill.com 最好不要直接QQ找我(拜托!)
iciq-lq@ln3:/THFS/home/iciq-lq$ cat /THFS/home/iciq-lq/bin/pot/Cu/POTCAR /THFS/home/iciq-lq/bin/pot/C/POTCAR /THFS/home/iciq-lq/bin/pot/H/POTCAR /THFS/home/iciq-lq/bin/pot/O/POTCAR > POTCAR iciq-lq@ln3:/THFS/home/iciq-lq$ grep TIT POTCAR TITEL = PAW_PBE Cu 22Jun2005 TITEL = PAW_PBE C 08Apr2002 TITEL = PAW_PBE H 15Jun2001 TITEL = PAW_PBE O 08Apr2002 iciq-lq@ln3:/THFS/home/iciq-lq$
#!/usr/bin/env bash # Create a GGA_PAW POTCAR file # by BigBro # To Use it: potcar.sh Cu C H O
# Define local potpaw_GGA pseudopotentialrepository: repo="/THFS/home/iciq-lq/bin/pot"
# Check if older version of POTCAR ispresent if [ -f POTCAR ] ; then mv -f POTCAR old-POTCAR echo" ** Warning: old POTCAR file found and renamed to 'old-POTCAR'." fi
# Main loop - concatenate the appropriatePOTCARs (or archives) for i in $* do iftest -f $repo/$i/POTCAR ; then cat $repo/$i/POTCAR>> POTCAR else echo" ** Warning: No suitable POTCAR for element '$i' found!! Skipped thiselement." fi done
iciq-lq@ln3:/THFS/home/iciq-lq$ ls bin LVASPTHW old-POTCAR POTCAR potcar.sh test_jobs iciq-lq@ln3:/THFS/home/iciq-lq$ potcar.sh Cu C H O ** Warning: old POTCAR file found and renamed to 'old-POTCAR'. iciq-lq@ln3:/THFS/home/iciq-lq$ ls bin LVASPTHW old-POTCAR POTCAR potcar.sh test_jobs iciq-lq@ln3:/THFS/home/iciq-lq$ grep TIT POTCAR TITEL = PAW_PBE Cu 22Jun2005 TITEL = PAW_PBE C 08Apr2002 TITEL = PAW_PBE H 15Jun2001 TITEL = PAW_PBE O 08Apr2002 iciq-lq@ln3:/THFS/home/iciq-lq$ iciq-lq@ln3:/THFS/home/iciq-lq$ potcar.sh BigBro ** Warning: old POTCAR file found and renamed to 'old-POTCAR'. ** Warning: No suitable POTCAR for element 'BigBro' found!! Skipped thiselement. iciq-lq@ln3:/THFS/home/iciq-lq$
当前目录下存在POTCAR,脚本会给出警告,将其重命名为old-POTCAR,然后生成新的POTCAR文件,包含Cu C H O 这四种元素。如果有一个元素不存在(上面实例总的BigBro),potcar库里找不到,则输出错误提示。
iciq-lq@ln3:/THFS/home/iciq-lq$ ls bin LVASPTHW test_jobs iciq-lq@ln3:/THFS/home/iciq-lq$ potcar.sh Cu_pv C H1.25 O iciq-lq@ln3:/THFS/home/iciq-lq$ ls bin LVASPTHW POTCAR test_jobs iciq-lq@ln3:/THFS/home/iciq-lq$ grep TIT POTCAR TITEL = PAW_PBE Cu_pv 06Sep2000 TITEL = PAW_PBE C 08Apr2002 TITEL = PAW_PBE H1.25 07Sep2000 TITEL = PAW_PBE O 08Apr2002 iciq-lq@ln3:/THFS/home/iciq-lq$
iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ potcar.sh $(head -n 6 POSCAR | tail -n 1) iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ grep TIT POTCAR TITEL = PAW_PBE Ru 04Feb2005 TITEL = PAW_PBE H 15Jun2001
大功告成!!!!
5.3 不想每次输入这么长的命令:可以将其写进 ~/.bashrc文件中:
1
alias pos2pot="potcar.sh $(head -n 6 POSCAR | tail -n 1)"
1 2 3 4 5 6
iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ ls POSCAR POTCAR iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ vi ~/.bashrc iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ . ~/.bashrc iciq-lq@ln3:/THFS/home/iciq-lq/LVASPTHW/potcar$ pos2pot ** Warning: old POTCAR file found and renamed to 'old-POTCAR'.
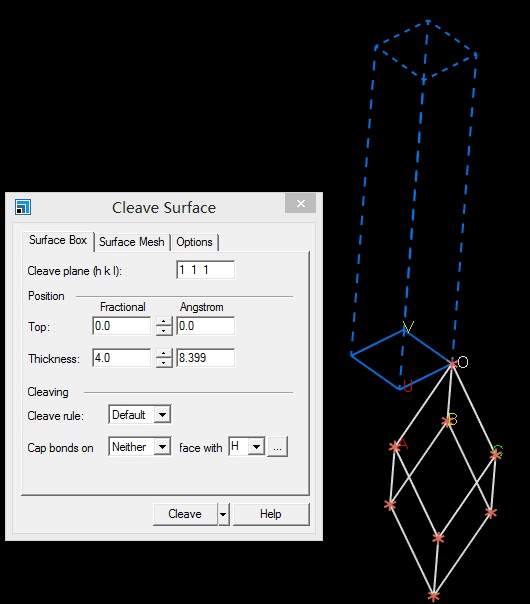
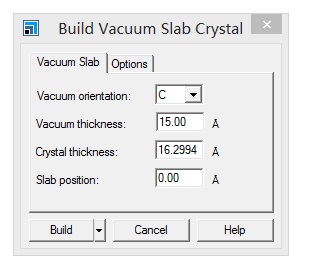


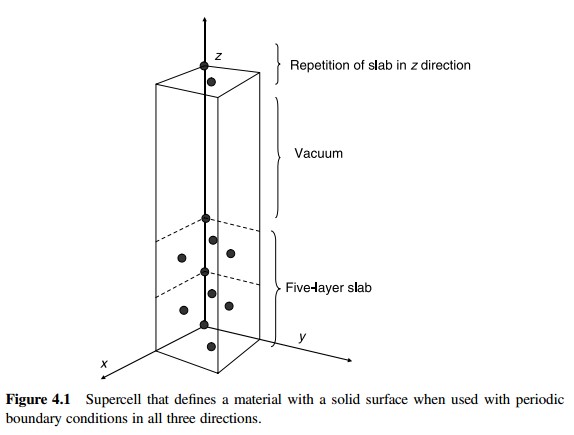
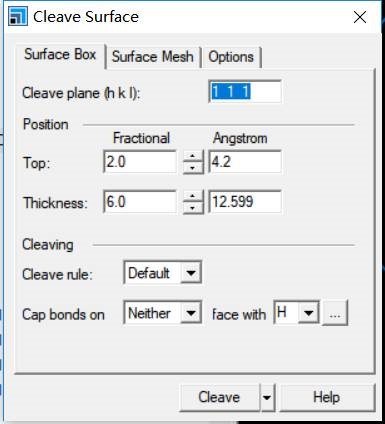
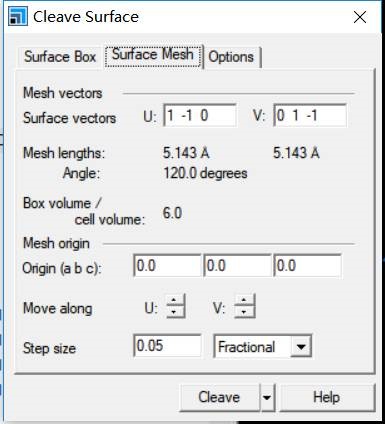
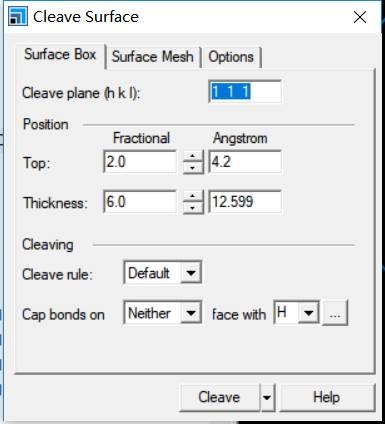
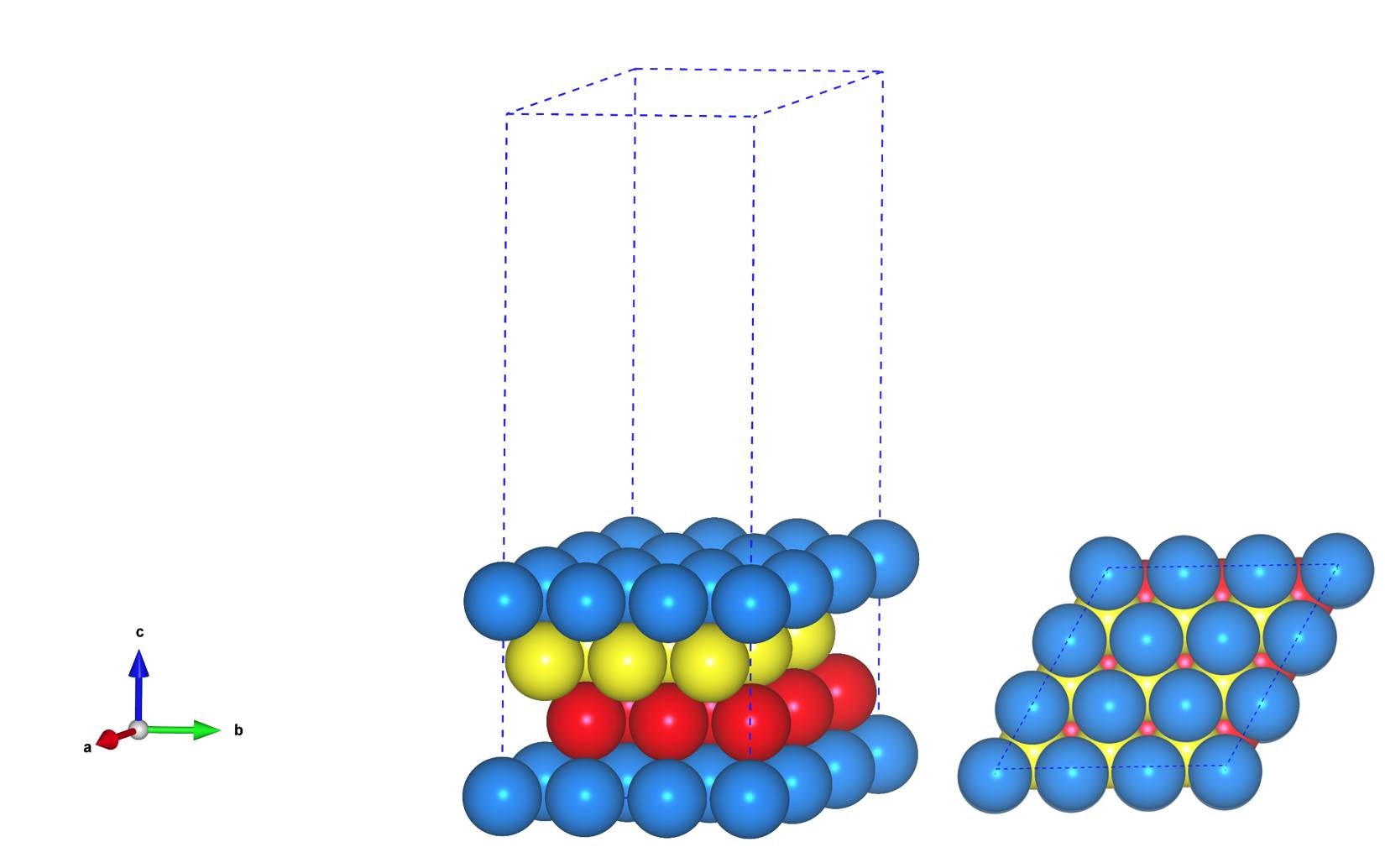 主要使用Material Studio进行切面操作,这一部分,本人不想做过多的解释,因为各大培训班,网上各种博客都有操作说明。本节大师兄通过Cu(111)面的例子给大家简单演示一下。
主要使用Material Studio进行切面操作,这一部分,本人不想做过多的解释,因为各大培训班,网上各种博客都有操作说明。本节大师兄通过Cu(111)面的例子给大家简单演示一下。