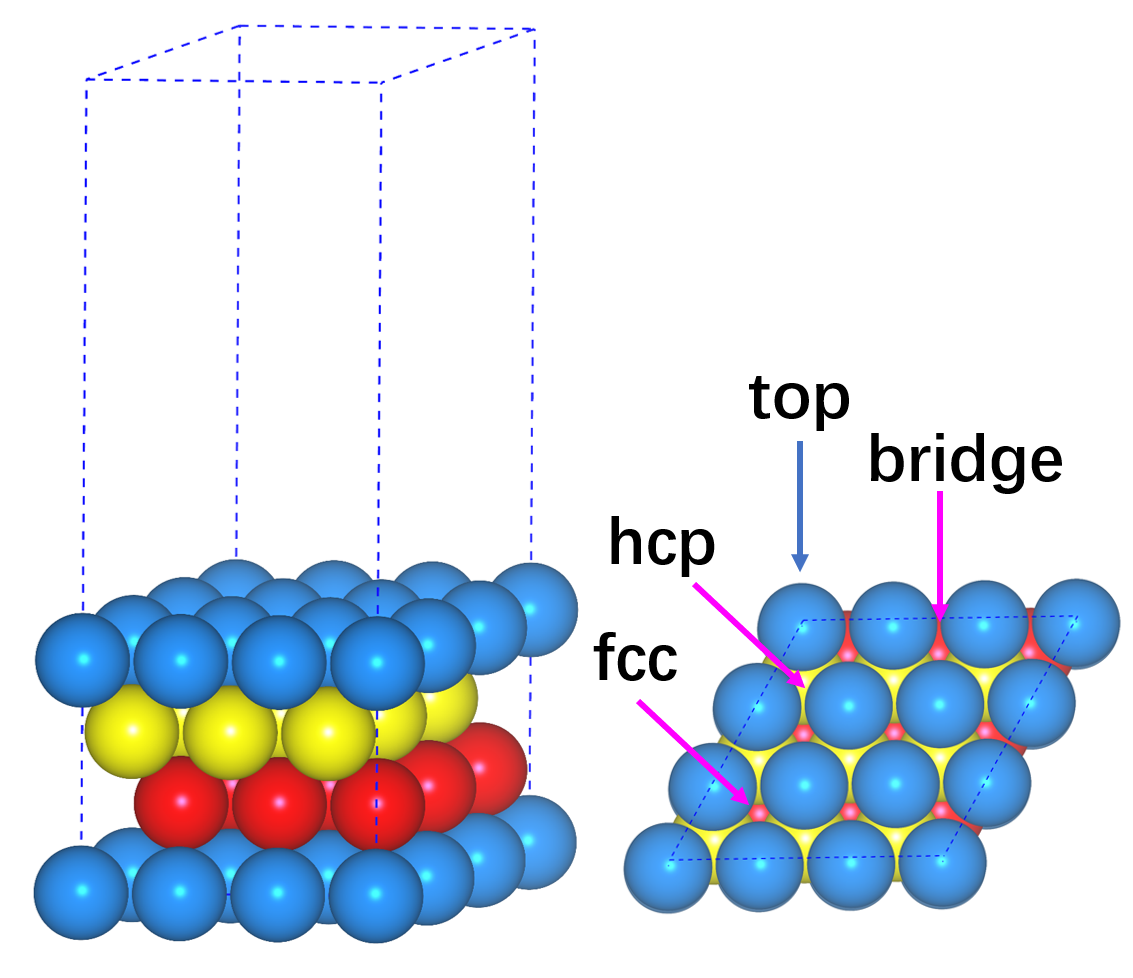
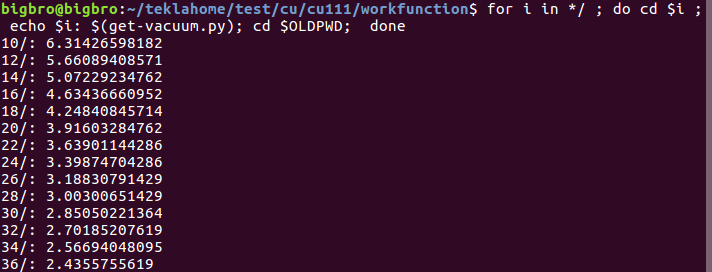
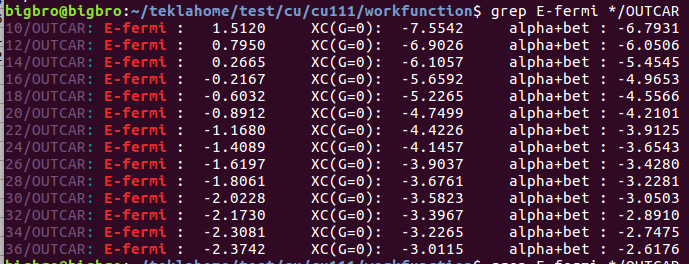
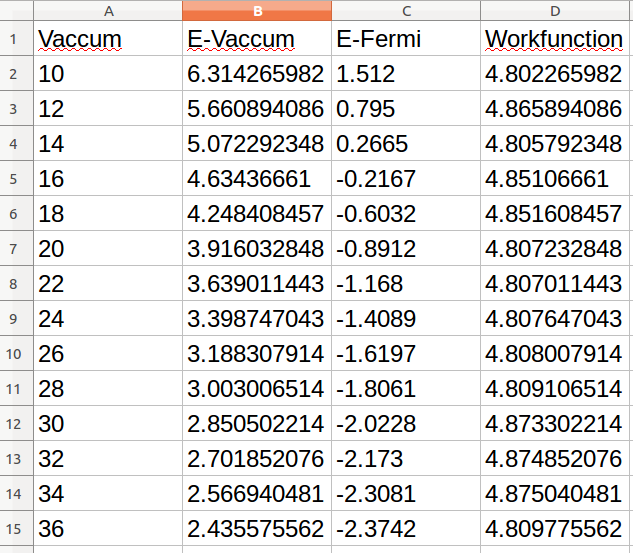
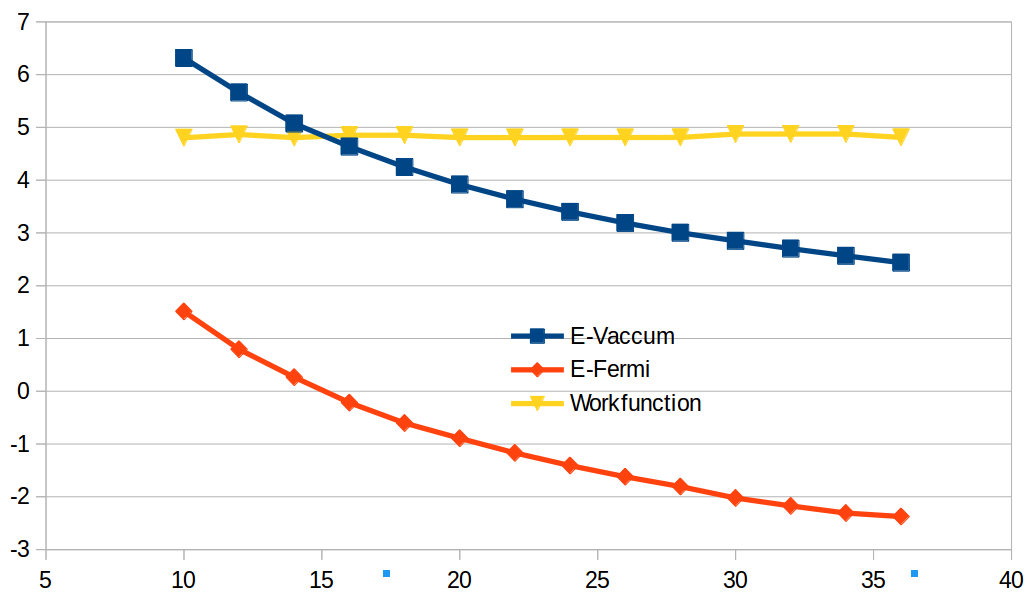
Ex63 吸附能的计算(八)
在进行本文的内容之前,大家先玩个人尽皆知的小游戏:
找不同
(一)找出下面两幅图中不同的部分。

相信这个大家很快都能找出来,找不出来的话,不建议继续浏览下面的部分。
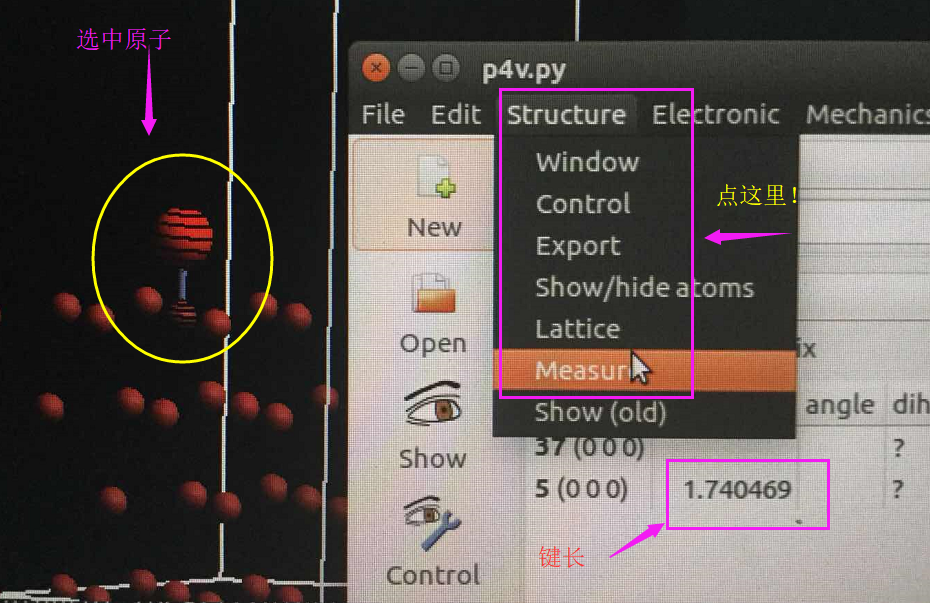
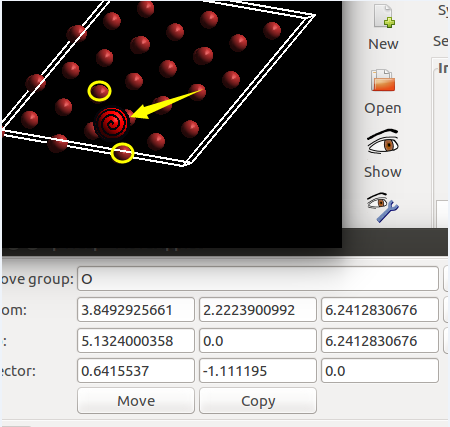
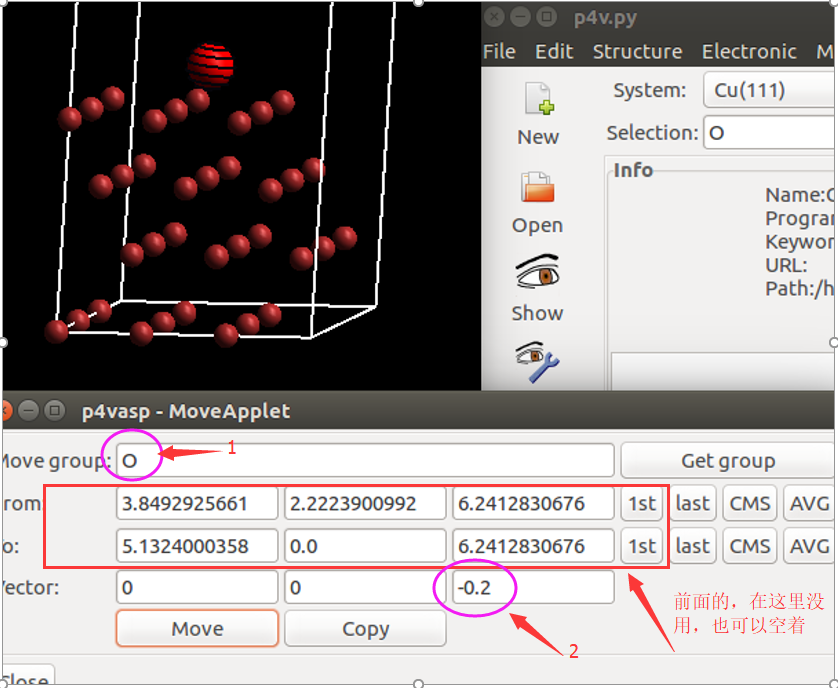
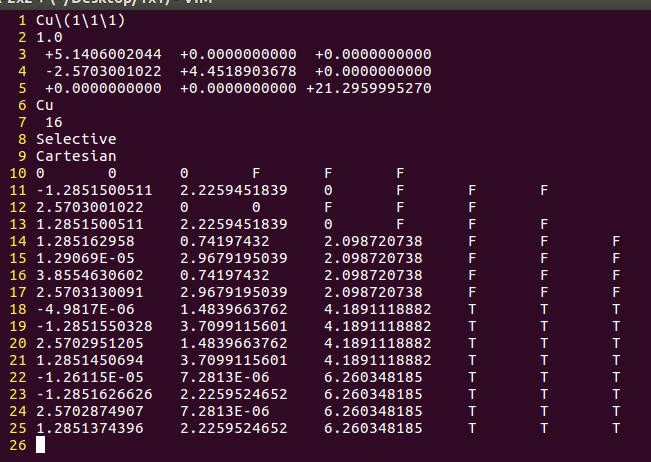
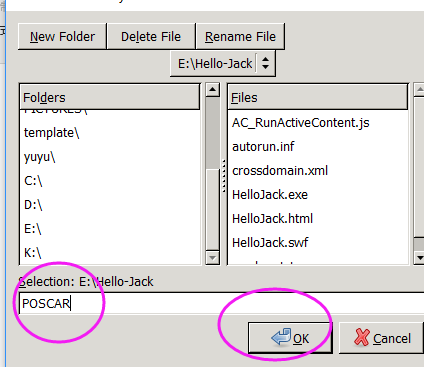
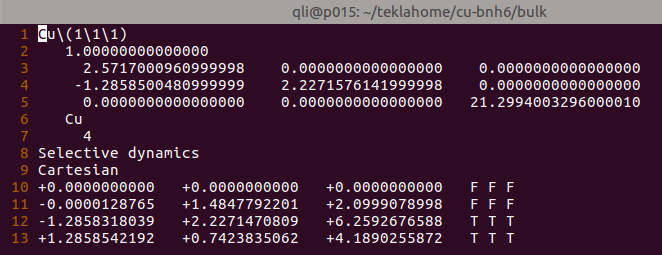
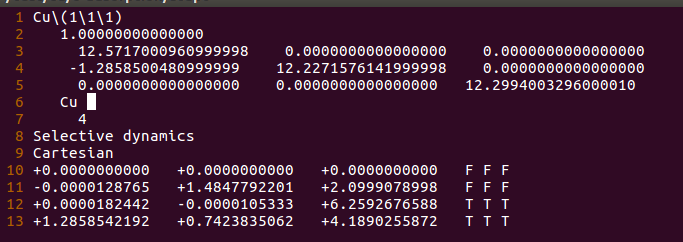
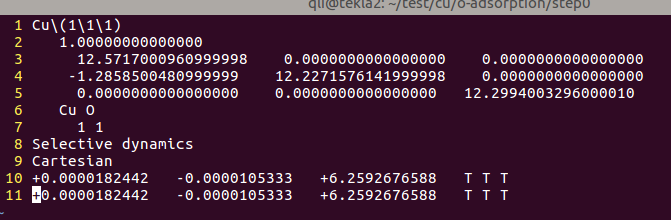
(二)下面两个截图中,找出不同的部分
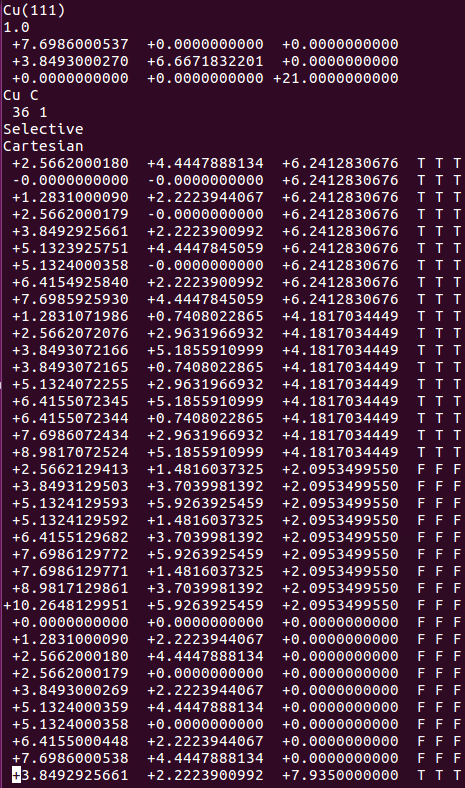
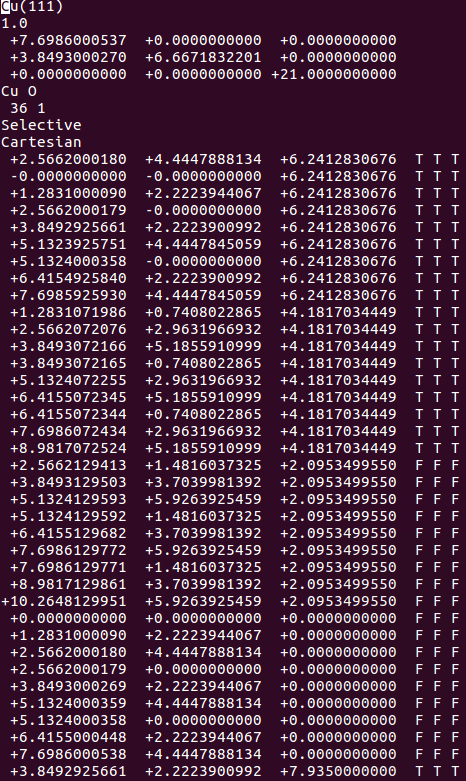
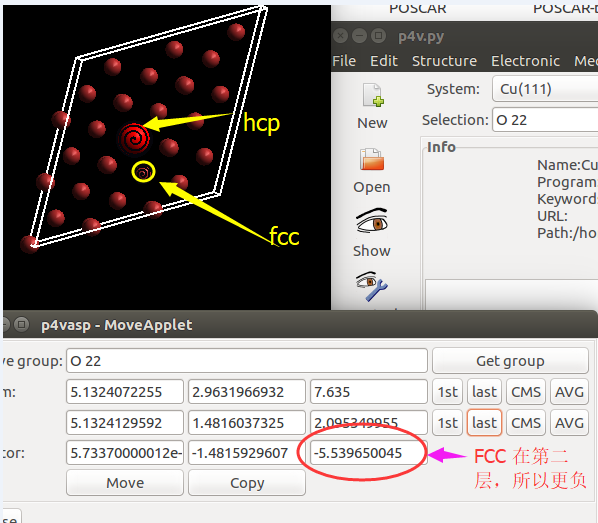
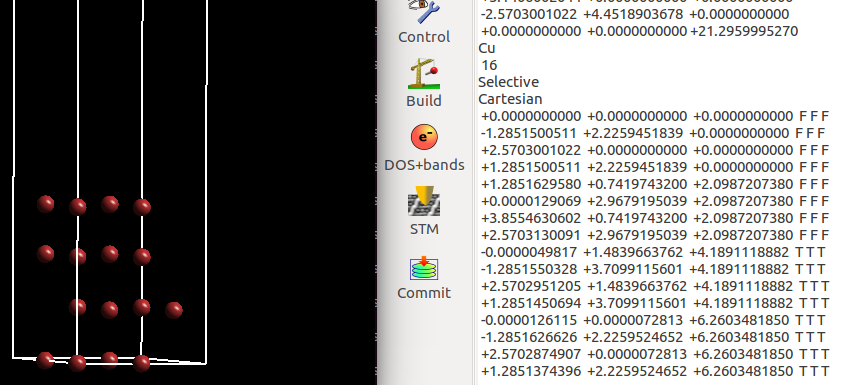
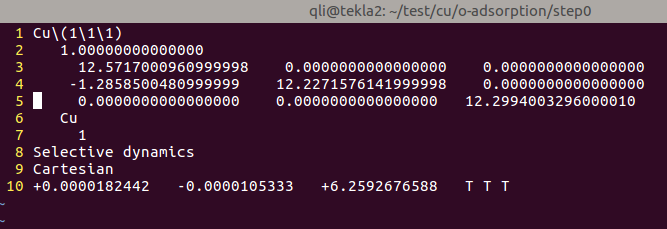
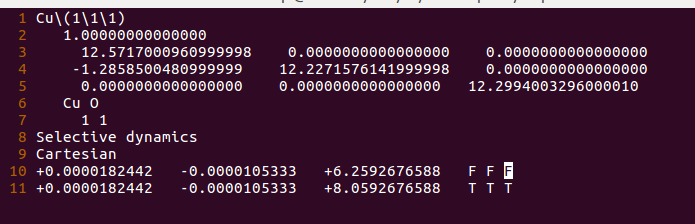
(三)下面两个截图中,找出不同的部分
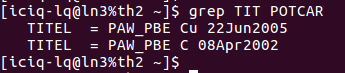
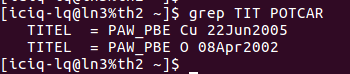
聪明美丽能干活泼善良可爱宇宙超级无敌的你会发现:
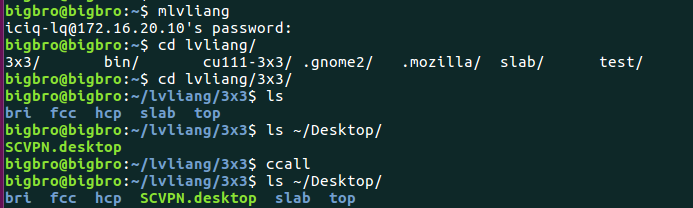
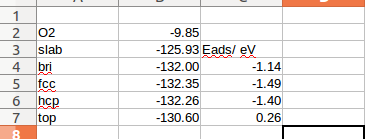
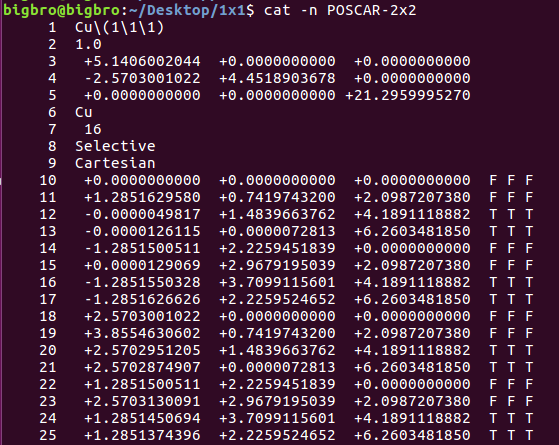
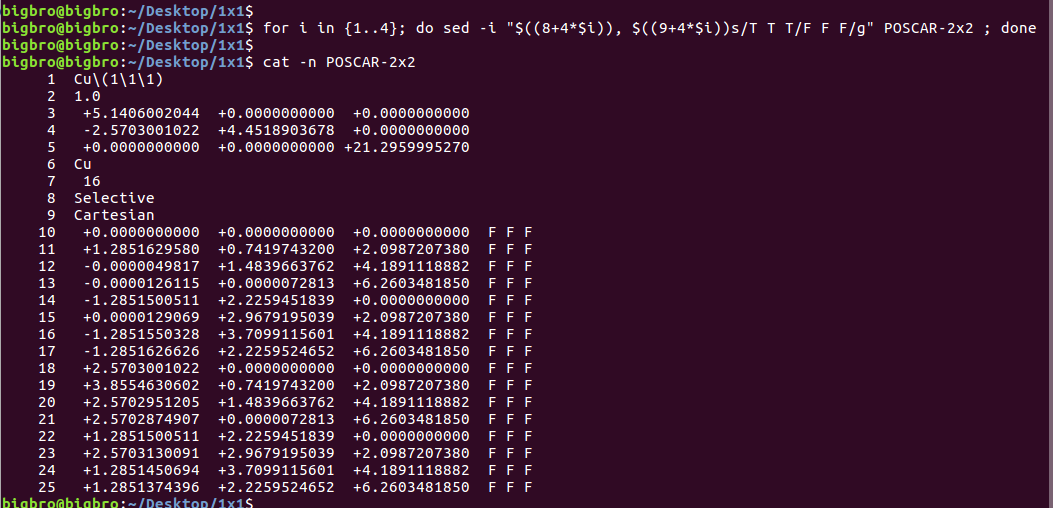
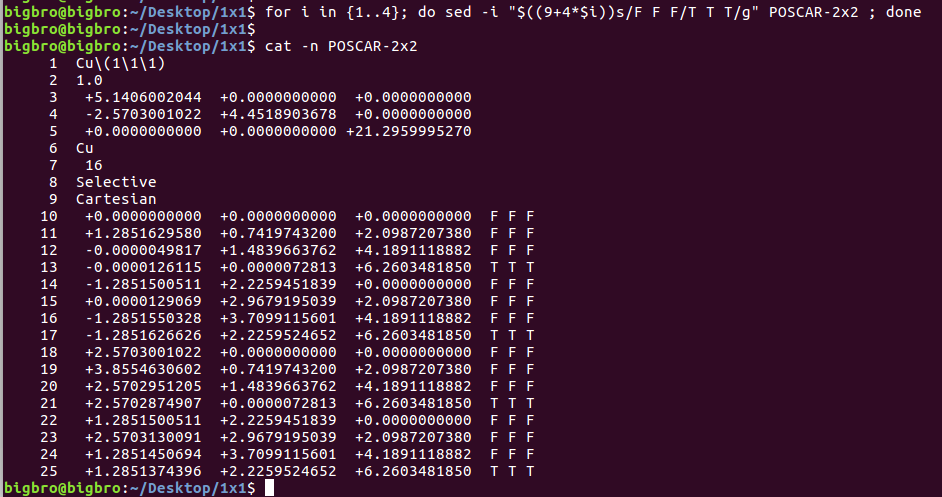
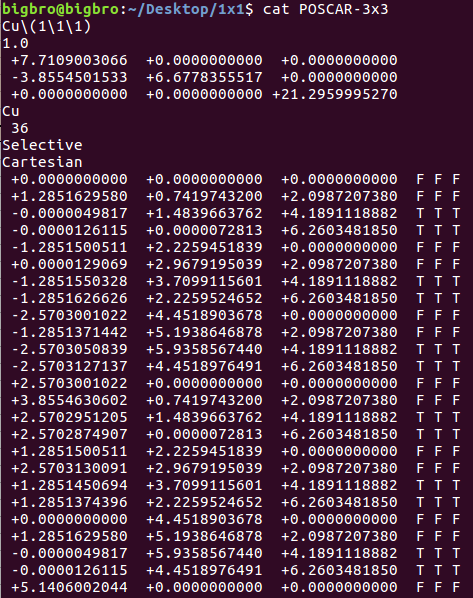
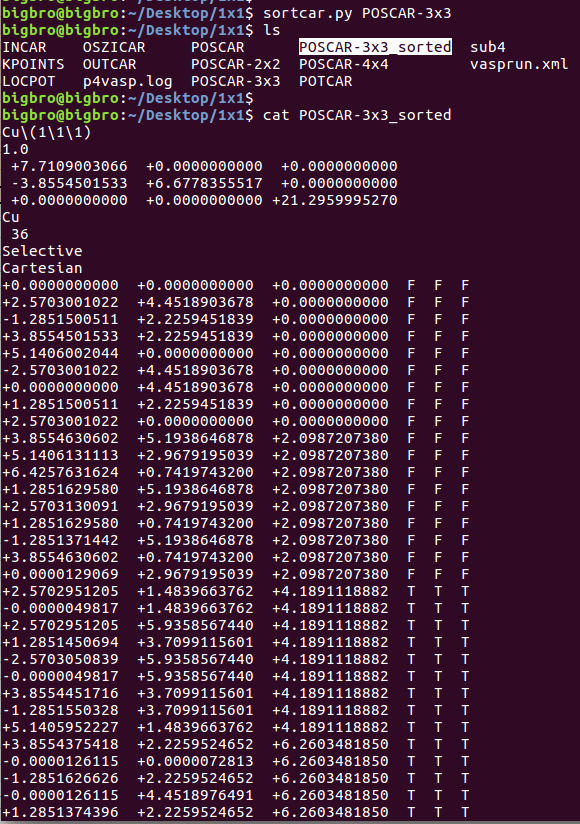
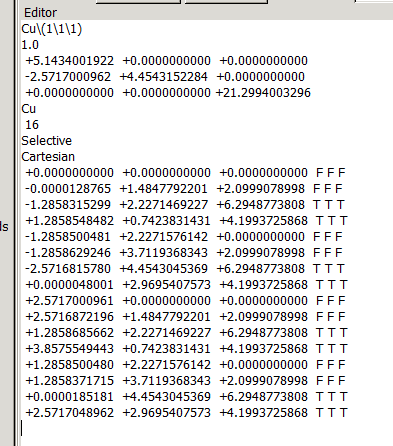
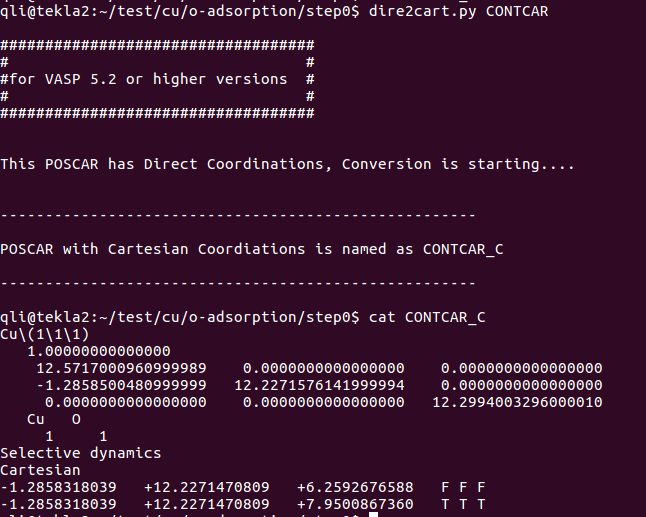
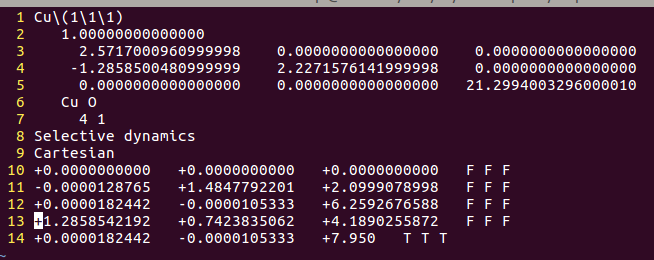
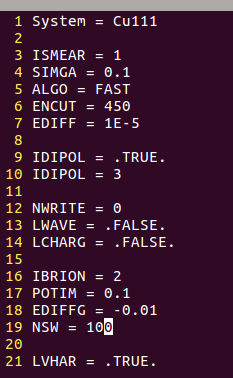
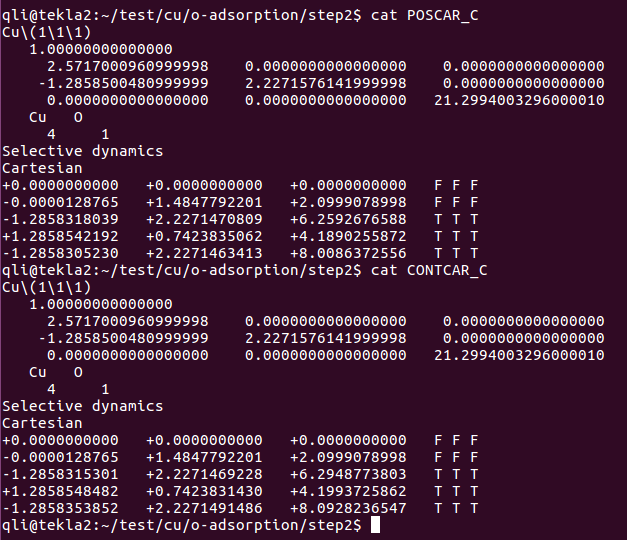
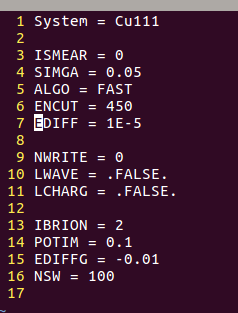
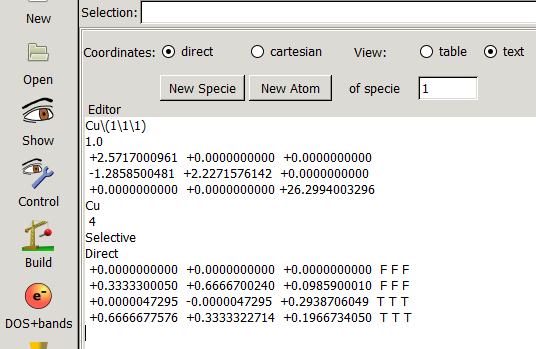
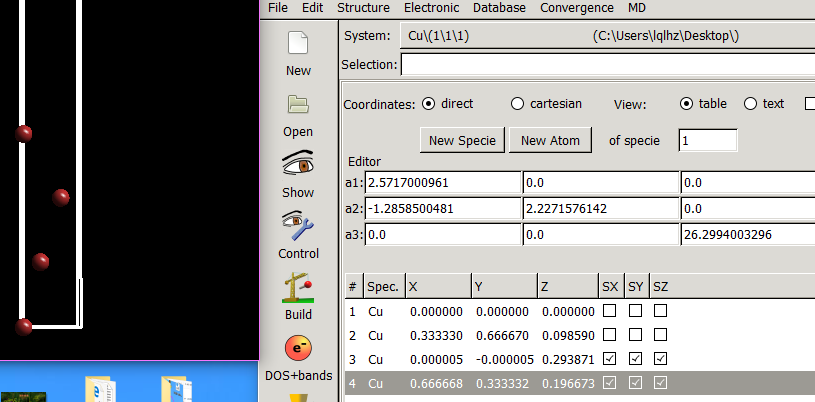
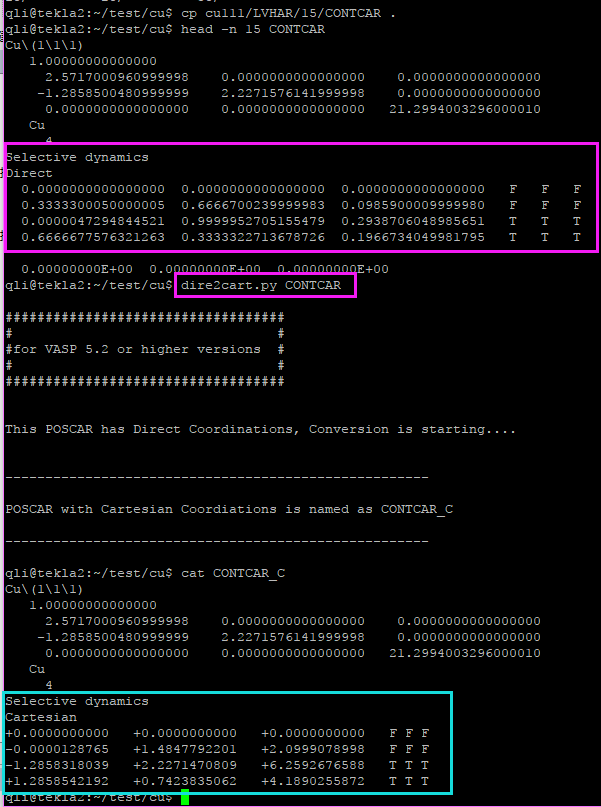
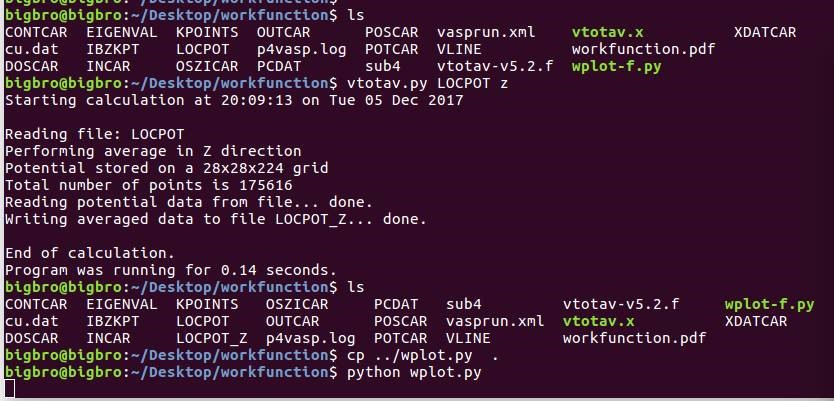
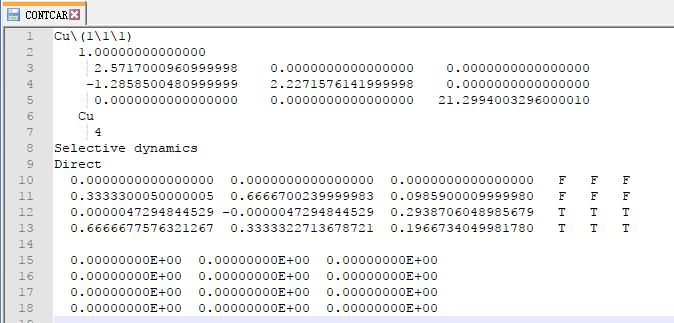
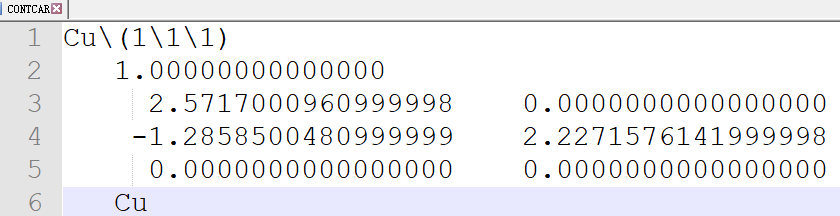
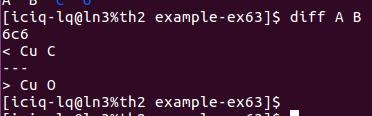
在找不同2中,两个图中内容的区别在于: POSCAR 的第6行
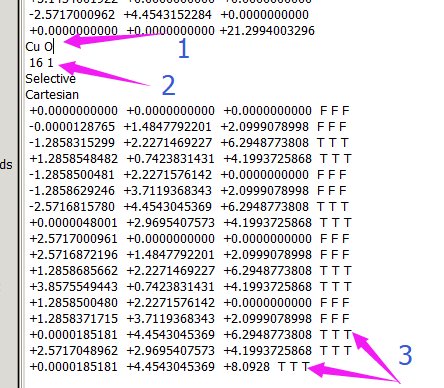
在找不同3中,两个图中内容的区别在于: POTCAR 的第二个元素
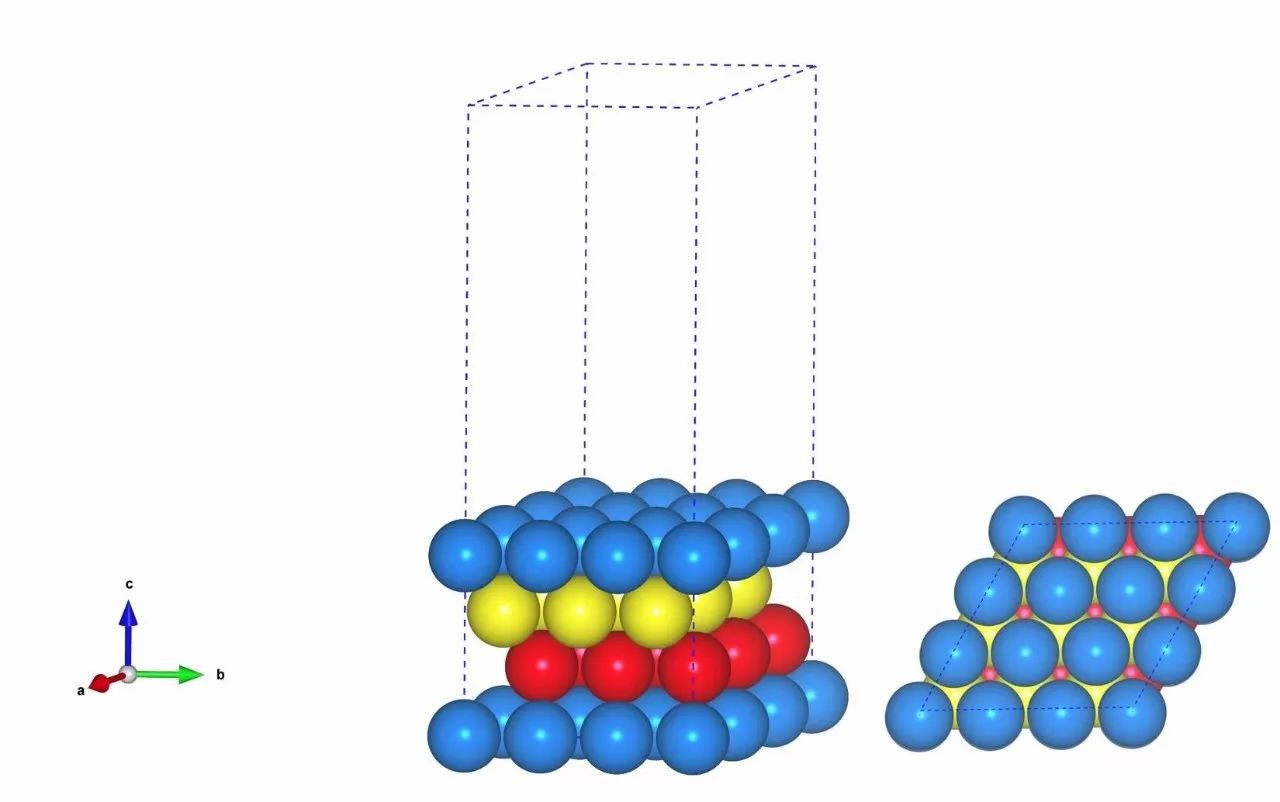
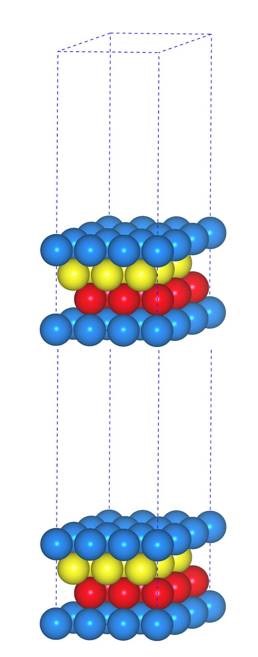
当你发现了这些不同之处,就应该差不多明白了,这是2个计算任务文件,一个是C原子在Cu(111), 另一个是O原子在Cu(111)上的计算。
仔细分析不同的地方,你就应该想到: 既然我们有了O吸附相关的计算文件,那么就可以在此模板基础上,快速编辑C的吸附模型,然后进行计算。
在进行下面的快速编辑模型的内容,先给大家介绍一个Linux下面常用的比较不同的命令:diff。简单操作如下,更高级的大家自由去百度学习,并加以发挥拓展。
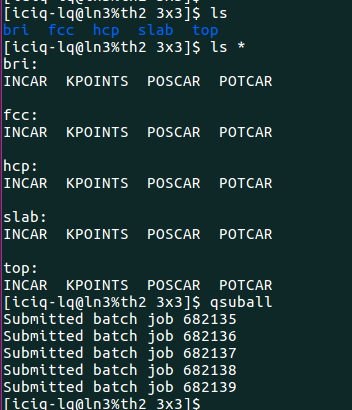
批量搭结构
快速准备C原子在Cu(111)上吸附的计算文件:
1)INCAR 保持一样
2)KPOINTS 保持一样
3)POSCAR: 将 O 直接替换成C就OK了。
i)直接编辑,无数种编辑器任你选。不过 Windows下一定要记得编辑完后运行下: dos2unix
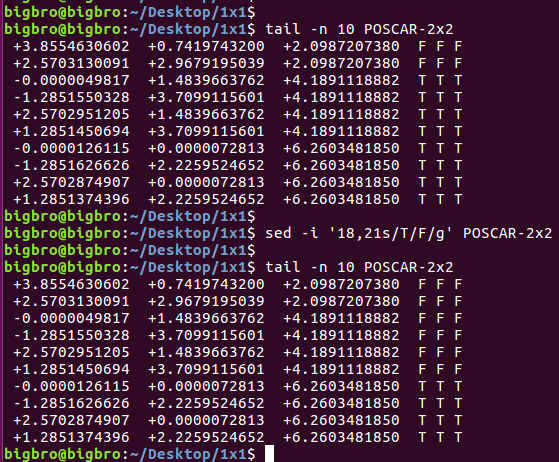
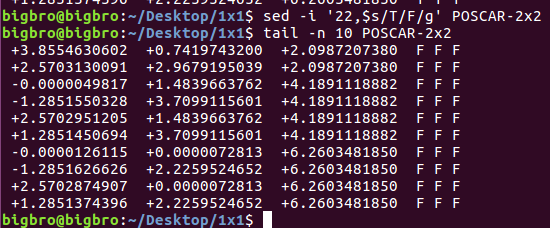
ii)使用sed命令: sed -i '6s/O/C/g' POSCAR
4)POTCAR:
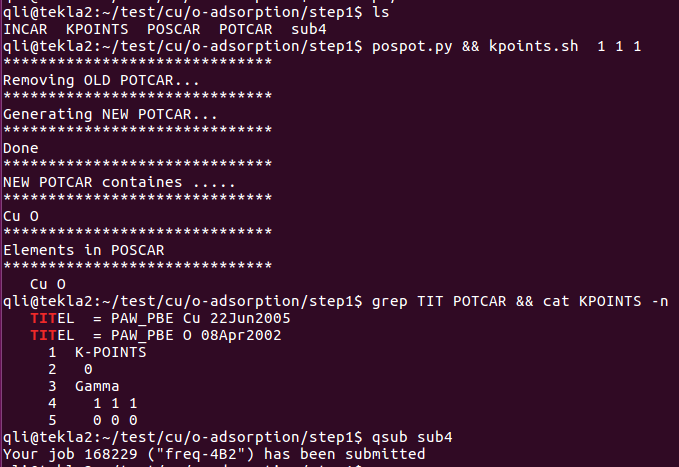
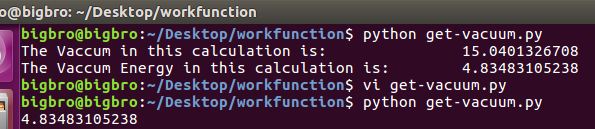
i) 使用前面我们介绍的脚本:potcar.sh, 脚本运行例子如下图:(去本书的附录章节里面找脚本)
https://www.bigbrosci.com/2017/12/21/A05/
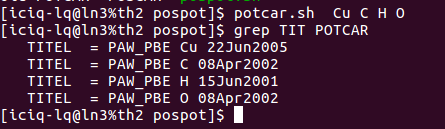
直接输入脚本名,后面跟你想要的元素名。
看下本人在天河II号服务器上的设置。
1) potcar.sh 在本人账号的: ~/bin 目录下
2) VASP所有的POTCAR文件在: ~/bin/pot 目录下
3) pot目录下,每个元素的文件夹中有2两个文件:POTCAR 和 PSCTR (PSCTR这个我也不知道有什么用。)
4)新手,没有经验,不会用超算的,不会Linux的,可以效仿下:

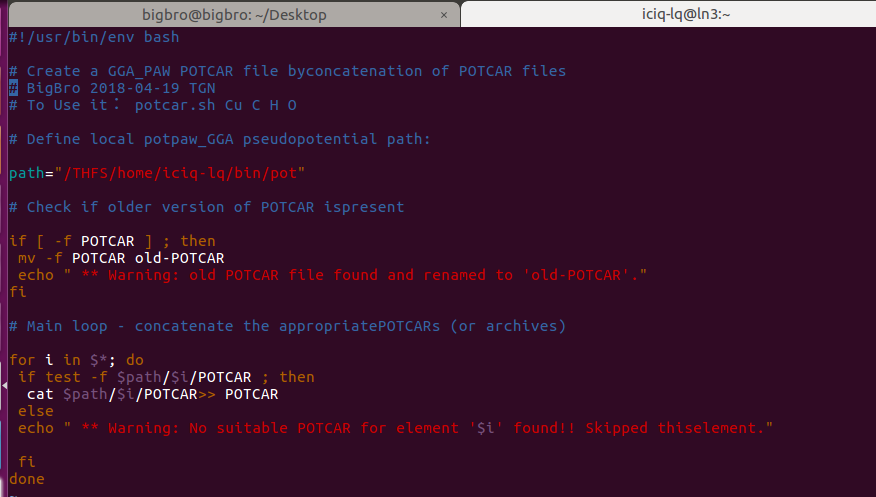
脚本下载后,将POTCAR所在的目录替换成你的: path=”你的目录”。
怎么知道目录呢? 使用上图中的pwd 这个命令。
ii) 我很多时候都懒得去看POSCAR中的元素以及顺序,便打算根据POSCAR中的内容生成对应的POTCAR,于是便写了一个脚本:pospot.sh。使用方法如下:
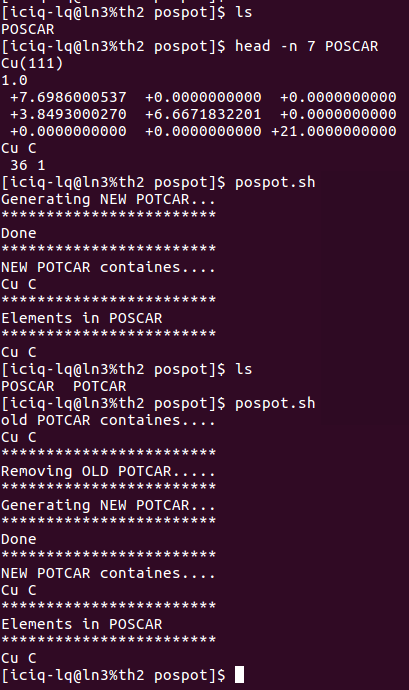
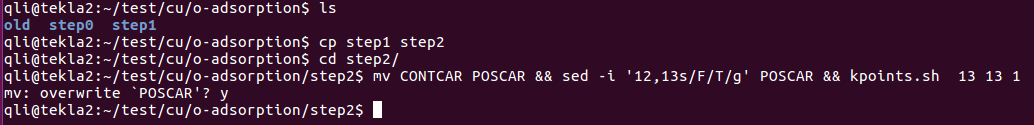
上图运行了这个脚本两次:
1) 第一次运行的时候,目录下没有POTCAR,于是脚本直接生成新的
2) 第二次运行的时候,目录下有了一个POTCAR,于是脚本将POTCAR中的元素顺序读出来,然后删掉旧的POTCAR,重复1)的操作,生成新的POTCAR。(大师兄本人的扯淡逻辑,大家可以忽略,脚本文章末尾下载直接使用即可。)
3) 注意:这个脚本读取的仅仅是以 元素 命名文件夹中的POTCAR。什么意思呢? 举个例子:Cu的POTCAR有:Cu,Cu_GW, Cu_pv,Cu_sv_GW这四种。脚本读取的仅仅是Cu中的POTCAR。如果你想用Cu_pv,那么请使用: potcar.sh Cu_pv C H O这个命令。
4) 脚本内容如下:大家可以自己手动敲一遍,找找感觉。
1 |
|
5)提交任务的脚本保持一样
前面所说的都是一些具体的做法,用化学的思维来说,应该叫基元反应。当你掌握了这些具体的做法后,可以通过批处理的方式将它们集成起来。本人的做法如下:
1)批量编辑POSCAR:
1 | sed -i '6s/O/C/g' */POSCAR |
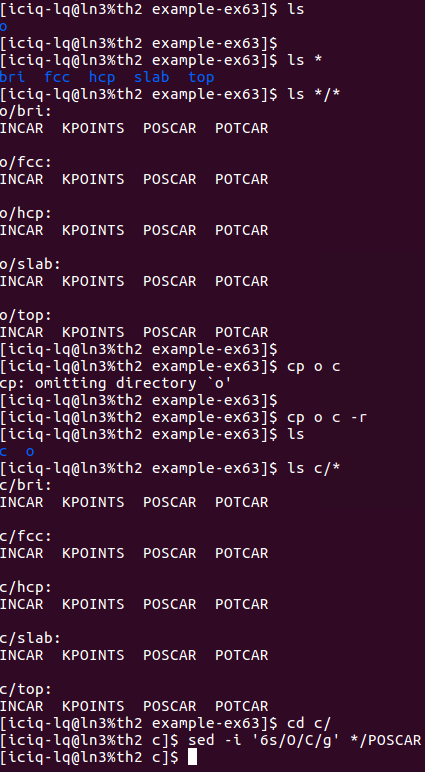
i) 将O原子的计算直接复制成C原子的
ii)使用sed 批量编辑POSCAR中的元素行。
2)完成前面的一步,直接批量提交任务: 提交任务的时候,脚本自动根据POSCAR生成对应的POTCAR.
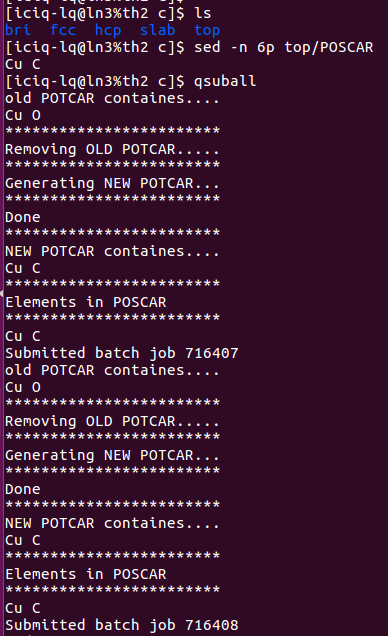
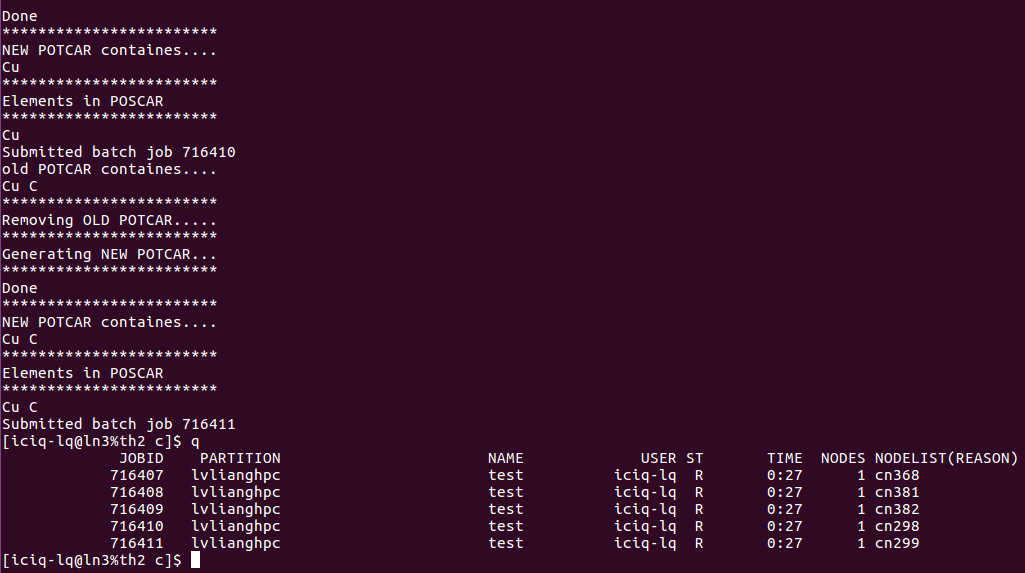
我是怎么做的呢?
在~/.bashrc 文件中 将pospot.sh 写到 qsuball里面了。大家可以看下我的~/bashrc文件:
1 | alias q='yhqueue' |
批量删除任务
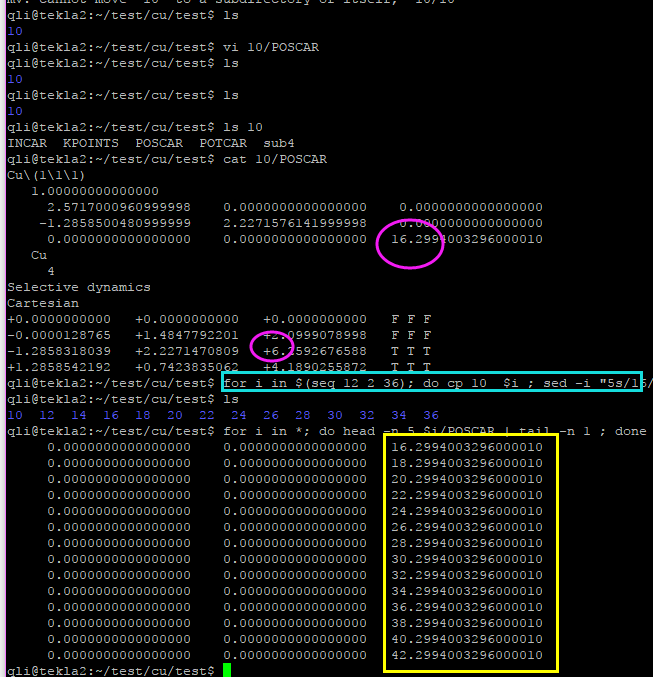
前面批量提交了任务,但这些任务已经算起来了,但这些结果对我没有用出,纯属浪费机时,所以我要杀死它们。这里介绍给大家一个简单的批量删除任务的命令: {..} 操作如下:
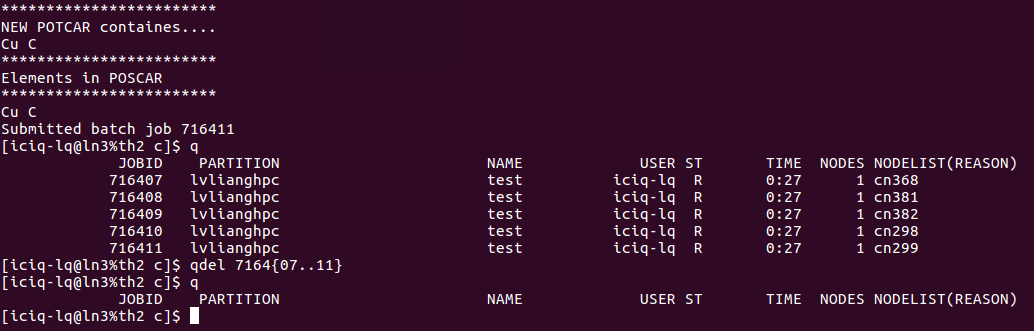
qdel 就是天河II号中的yhcancel命令,本人根据自己的习惯,修改了一下alias中的内容(见前面~/bashrc文件的内容)
如果你的任务不是连续的,也可以使用这个命令, 只要把第一个和最后一个任务的ID号写在花括号里两个点..的两端即可。非常简单,暴力和直接。
中间如果有些任务不是你的,即使你想杀死也没有权限,系统会报错,直接忽视即可。
找不同
前面我们弄完了C原子的计算任务,下面我们再玩一次找不同的游戏
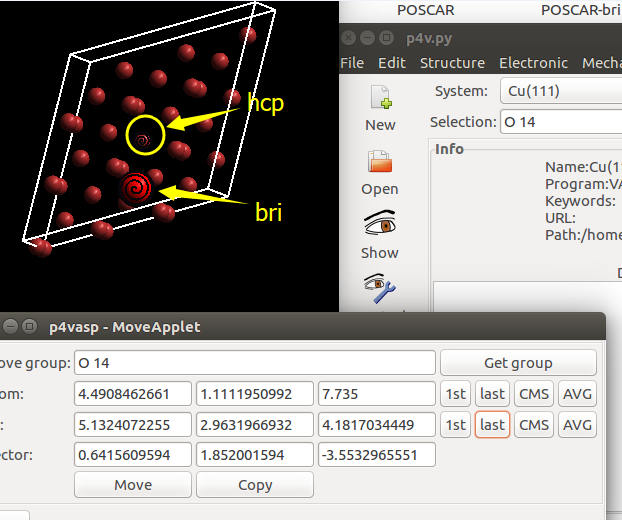
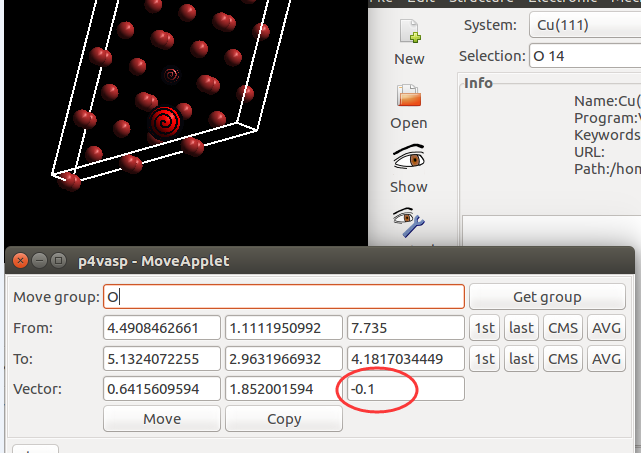
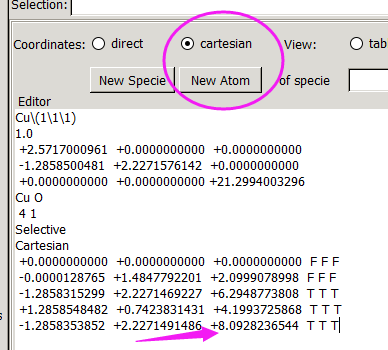
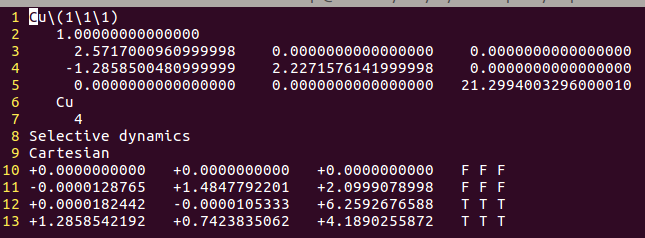
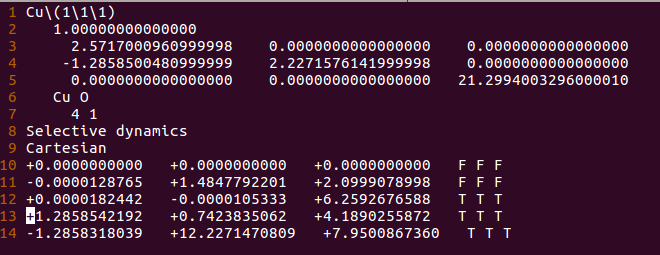
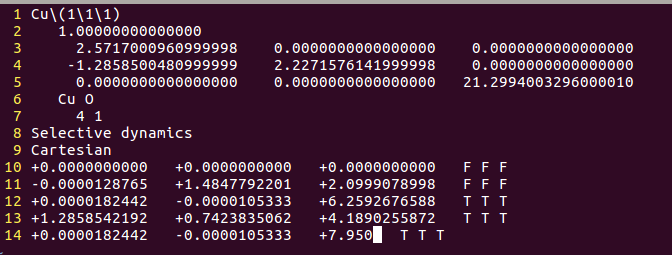
找不同4:下面2个截图中找出不同的地方:
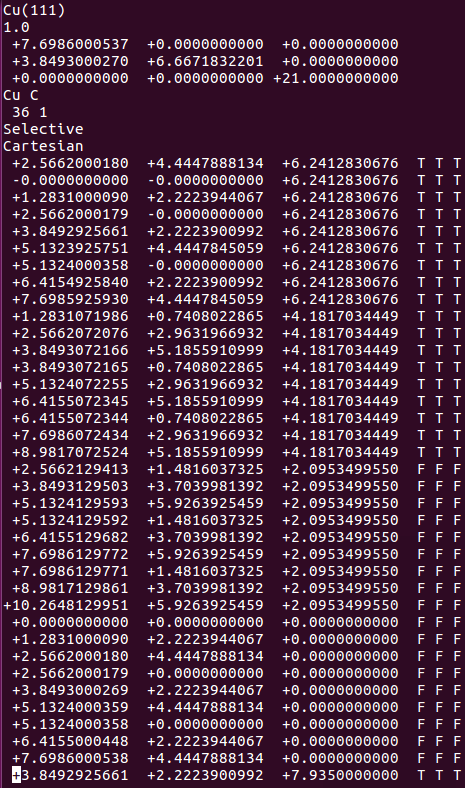
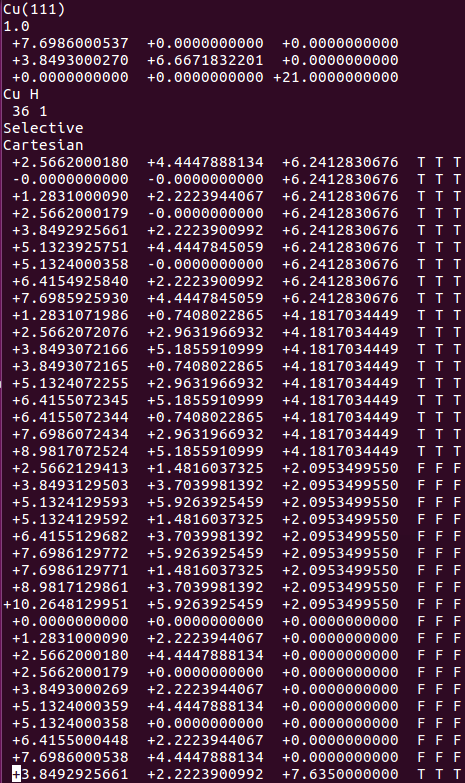
师兄,这个题很简单,你要算H原子的吸附。跟前面的操作是一样的。
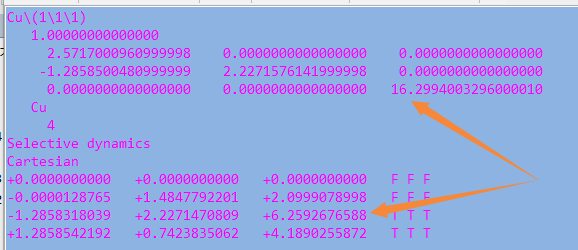
但是,请看最后一行。
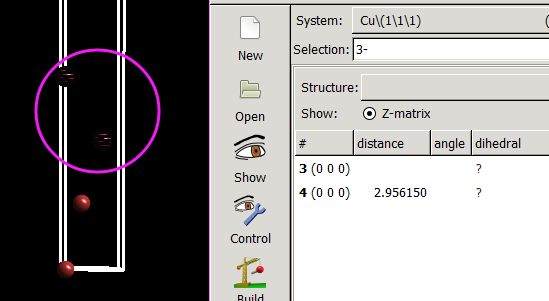
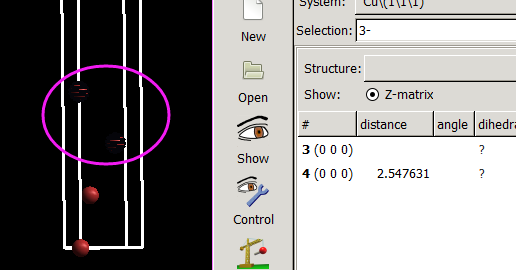
仔细的你会发现,这个题跟前面的有些不一样,原因在于最底部的坐标也稍微变化了一下,从7.935变成 7.635了。 这是为什么呢?
原因在于和C,O原子比起来,H原子很小,它在表面上吸附的时候,会更贴近表面。因此我们把z方向的坐标稍微修改了一下: 减小了 0.3 A。这样做的话可以保证我们的初始结构更具有合理的物理化学意义,从而加快计算的收敛,节约时间。大家在搭建模型的时候,结构合理是必须要考虑的,也是脑子里要时刻思考的事情。
通过sed命令批量操作在H原子的吸附上,只能完成一半的任务,也就是把元素行中的C或者O改成H。而另一半的任务:坐标的修改,则需要大家自己手动操作一下(记得在Cartesian坐标下修改,如果你脑子转的很快,可以迅速将Direct转化为Cartesian的话,也可以直接编辑Direct坐标)。
扩展练习:
1) 完成本节的所有操作;
2) 计算C,H,O在Cu(111)表面上,不同位点上的吸附;
3) 思考脚本的运行原理;
4) 思考如何通过:(找不同 + 写脚本)来节省自己的体力;
5) 思考自己平时常见的人工错误,并尝试用脚本来解决或者避免。
总结:
本文的题目是闭着眼算,不是让你真正闭着眼去提交任务,而是不通过可视化的界面进行结构的批量搭建,输入文件的批量处理以及提交任务。希望大家在搭结构的时候,多一些思考,少一些操作,在搭建合理结构的路上突飞猛进。