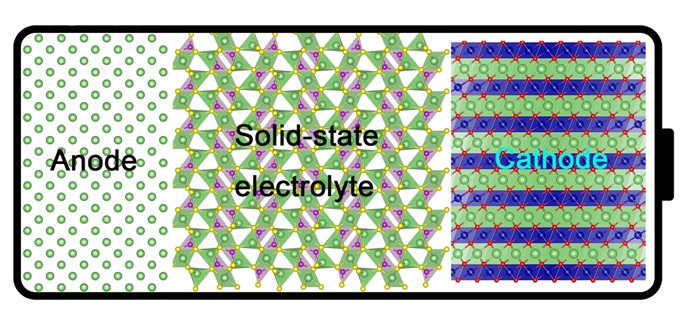
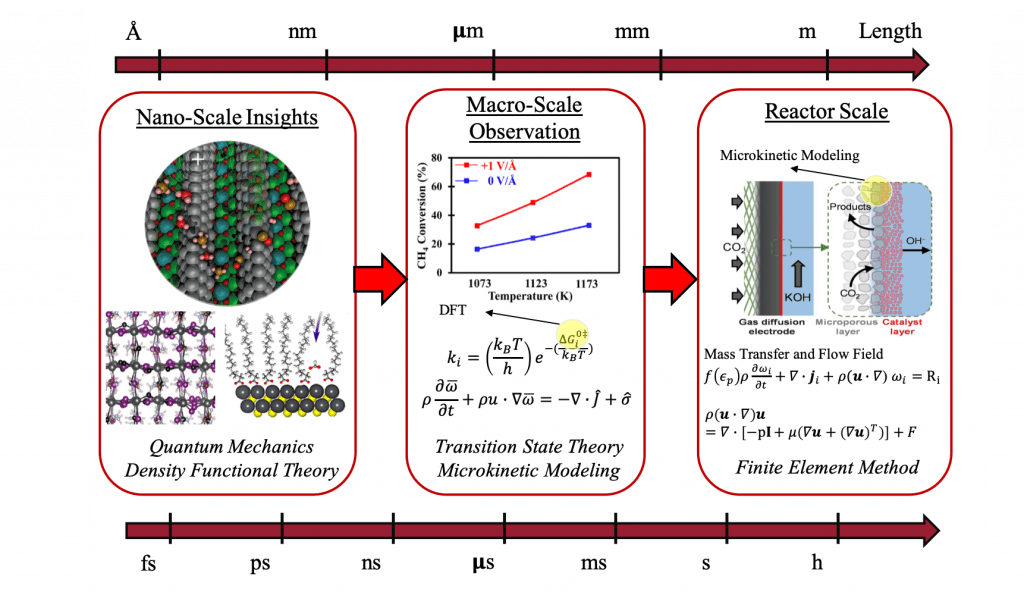
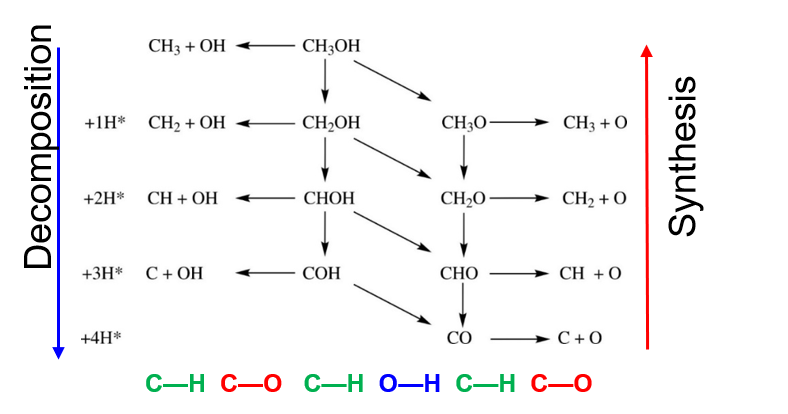
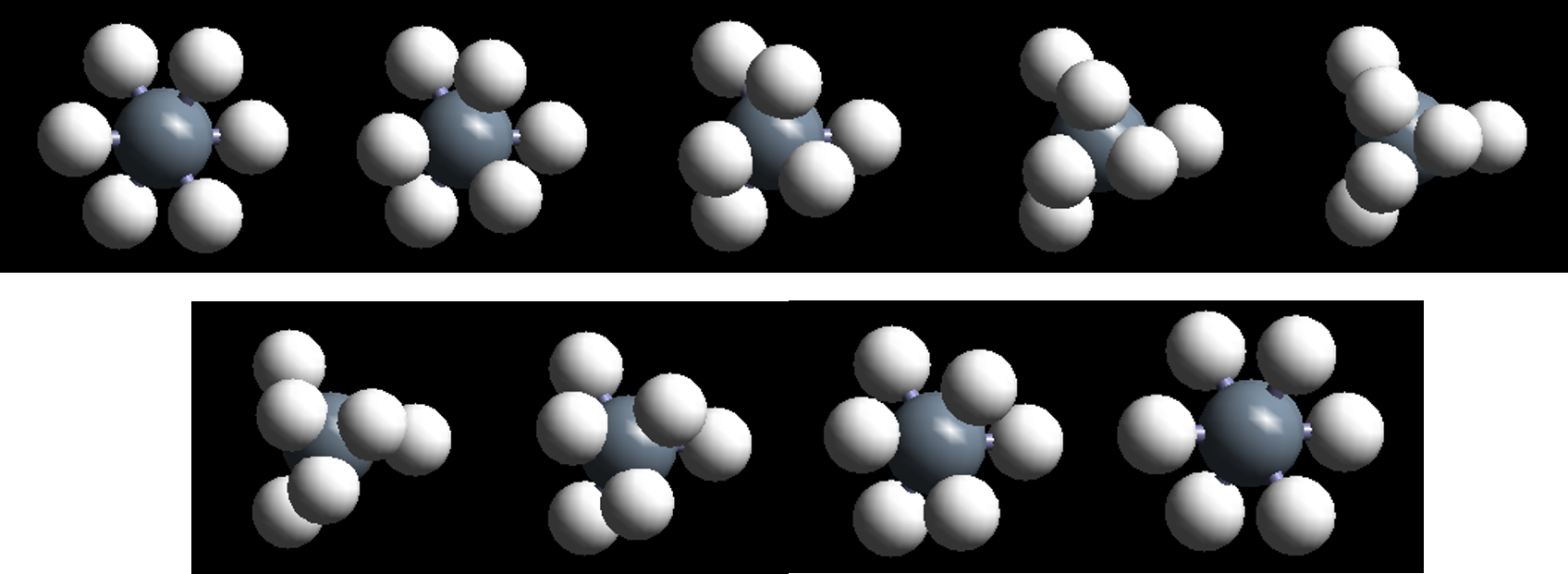
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
|
from ase import Atoms
import ase.io
from ase.build import molecule
from ase.build import bulk
from ase.build import surface
from ase.build import add_vacuum
from ase.build import fcc111, bcc110, hcp0001
from ase.constraints import FixAtoms
import subprocess
bcc = ['V', 'Cr', 'Mn', 'Fe', 'Nb', 'Pb']
hcp = ['Mg', 'Sc', 'Ti', 'Co', 'Zn', 'Y', 'Zr', 'Tc', 'Ru', 'Cd', 'Hf', 'Re', 'Os']
fcc = ['Al', 'Ca', 'Ni', 'Cu', 'Rh', 'Pd', 'Ag', 'Ir', 'Pt', 'Au']
dict_metals = {
'Ag':(-10.88004463,4,4.1423472817),
'Co':(-14.06155869,2,2.4908062578,4.0275560997),
'Cu':(-14.91182926,4,3.6339719976),
'Fe':(-16.47105782,2,2.8346922247),
'Ir':(-35.00402169,4,3.8852086642),
'Ni':(-21.86901226,4,3.5177809803),
'Pd':(-20.864555,4,3.9374172967),
'Pt':(-24.39436715,4,3.9669414218),
'Rh':(-29.10896058,4,3.8241655305),
'Ru':(-18.49439863,2,2.7126893229,4.2897522328),
}
def bottom(file_in):
'''This function is used to pull the cetered atoms (from ASE) back to the bottom. '''
f = open(file_in, 'r')
lines = f.readlines()
f.close()
coord = [float(line.rstrip().split()[2]) for line in lines[9:]]
bottom = min(coord)
out_put = open(file_in + '_bottomed', 'w')
out_put.writelines(i for i in lines[0:9])
for line in lines[9:]:
infor = line.rstrip().split()
infor[2] = str(float(infor[2]) - bottom)
out_put.write(' '.join(infor) + '\n')
out_put.close()
def cssm(metal, data_dict):
name = 'POSCAR_' + metal
if metal in bcc:
lattice_a = float(data_dict.get(metal)[2])
for i in range(1,4):
name_out = name + '_' + str(i)
slab = bcc110(metal, a = lattice_a, size = (i, i, 4), vacuum = 7.5)
'''(i,i,4) means repeat i i 4 in x y and z directions. vacuum will be 7.5 * 2 because it was added on the two sides.'''
constraint_l = FixAtoms(indices=[atom.index for atom in slab if atom.index < i*i*2])
slab.set_constraint(constraint_l)
ase.io.write(name_out, slab, format='vasp')
subprocess.call(['sed -i ' + '\'5a' + metal + '\' ' + name_out], shell = True )
bottom(name_out)
elif metal in hcp:
lattice_a, lattice_c = [float(i) for i in data_dict.get(metal)[2:]]
for i in range(1,4):
name_out = name + '_' + str(i)
slab = hcp0001(metal, a = lattice_a, c = lattice_c, size = (i, i, 4), vacuum = 7.5)
constraint_l = FixAtoms(indices=[atom.index for atom in slab if atom.index < i*i*2])
slab.set_constraint(constraint_l)
ase.io.write(name_out, slab, format='vasp')
subprocess.call(['sed -i ' + '\'5a' + metal + '\' ' + name_out], shell = True )
bottom(name_out)
elif metal in fcc:
lattice_a = float(data_dict.get(metal)[2])
for i in range(1,4):
name_out = name + '_' + str(i)
slab = fcc111(metal, a = lattice_a, size = (i, i, 4), vacuum = 7.5)
constraint_l = FixAtoms(indices=[atom.index for atom in slab if atom.index < i*i*2])
slab.set_constraint(constraint_l)
ase.io.write(name_out, slab, format='vasp')
subprocess.call(['sed -i ' + '\'5a' + metal + '\' ' + name_out], shell = True )
bottom(name_out)
else:
print('Please add your element in the crystal structure lists: bcc, hcp, and fcc')
for metal in dict_metals.keys():
cssm(metal, dict_metals)
|
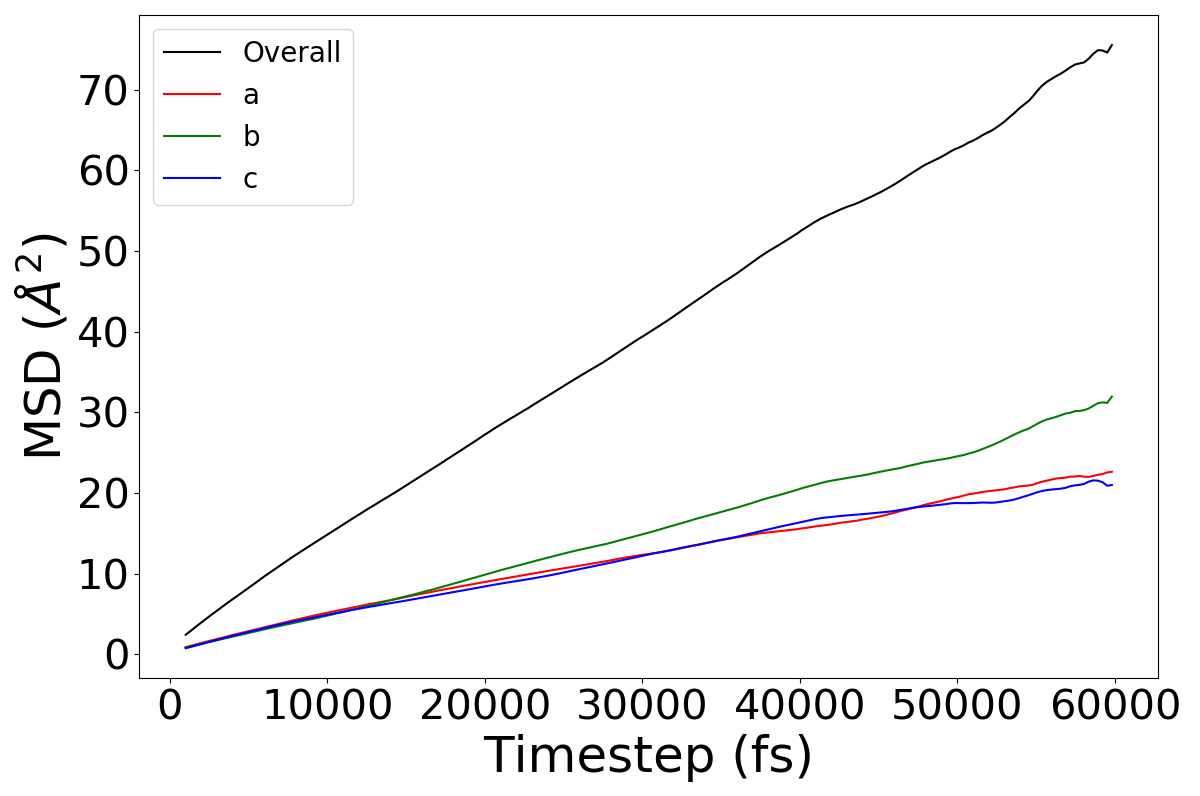

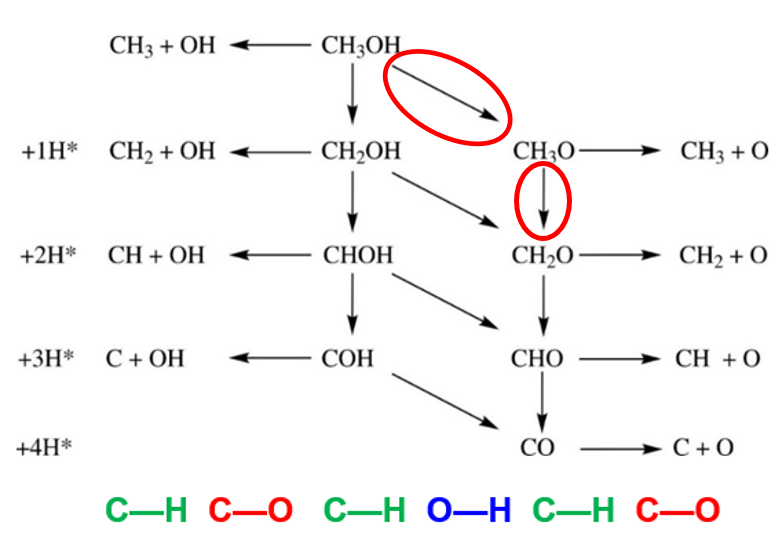
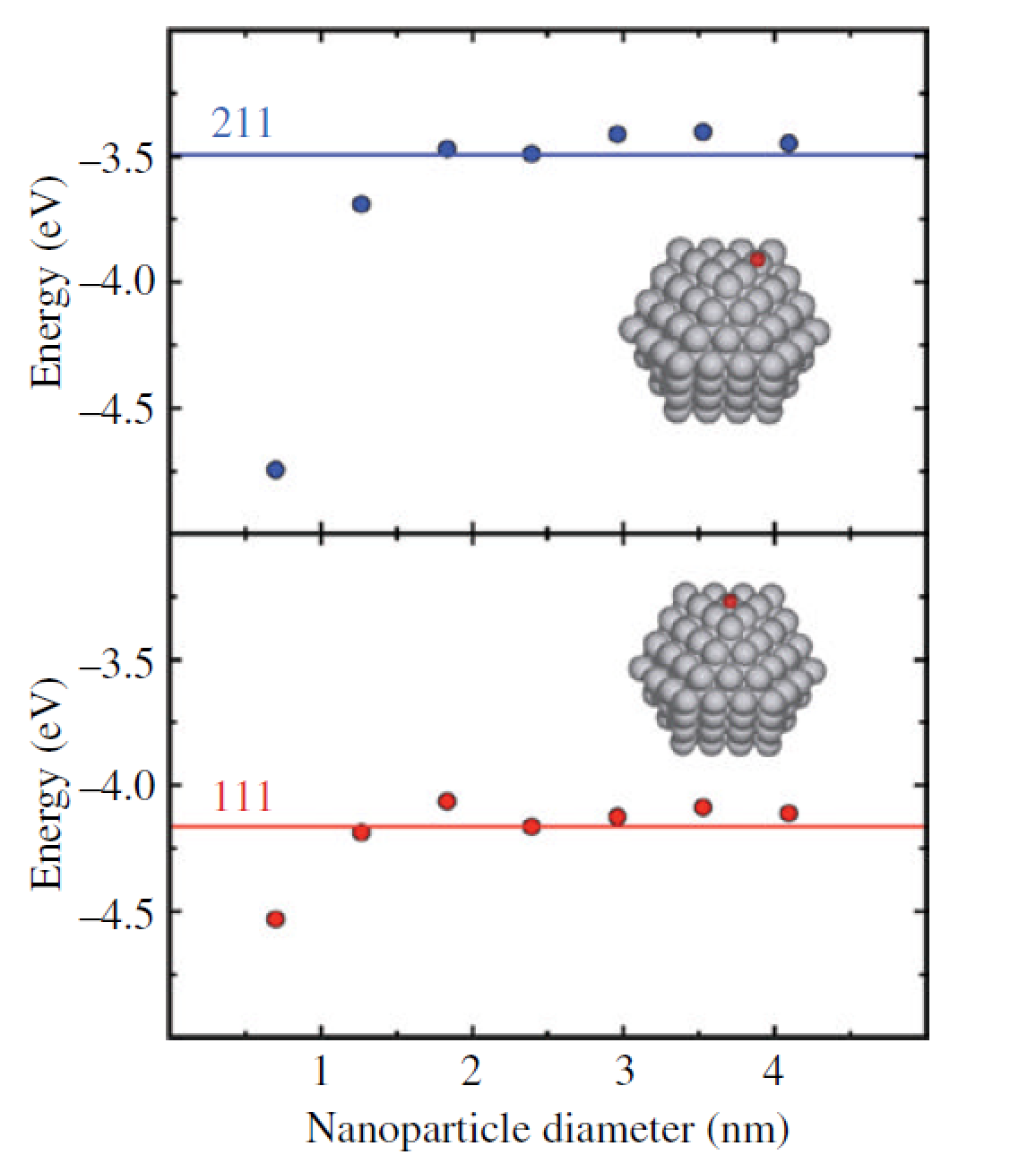


















 **
**