Ex80 nebmake.pl 的坑(一)
算过渡态,光知道检查虚频是不对的。如果算出来虚频多,很大程度是因为你的NEB初始结构有问题,也就是说你的过渡态路径背后的物理化学意义不是那么地理想。而对于NEB的初始结构,大部分人都是通过VTST的nebmake.pl脚本来实现的。
1 | nebmake.pl IS FS 8 |
分析上面这个命令:有4个关键的信息:
- 脚本:nebmake.pl
- 初始结构: IS
- 末态结构: FS
- 插的点数:8
本节默认你优化好了IS,FS,并且插8个点,分析一下脚本nebmake.pl的2个坑。其实,在Density Functional Theory: A Practical Introduction 这本书的第6章,其中的一个坑已经讲过了。强烈建议新手,老手认真看看这一章。
想知道这两个坑,首先我们先分析下nebmake.pl的工作原理。简举个化的一维例子:一个线段两端的坐标是x1和x2,我们把这个线段分成n份,并获得每一段的起始点的坐标。想必大家都知道怎么弄。
x_i = x0 + i *(x2-x1)/(n+1)
如果扩展到三维的xyz坐标,分别对y、z进行同样的处理。我们就得到了初始结构和末态结构之间这些的IMAGES的坐标。具体见:第六章的148页。

知道了原理,现在就可以分析其中的两个坑了。
坑1
前面讲的原子在表面扩散的例子,如果原子从fcc扩散到hcp。fcc和hcp的结构中,原子在z方向的坐标基本是相同的。如果运行nebmake.pl 这个命令,就会出现这样的一种情况,所有的IMAGE结构中,z的坐标几乎不变,也就是原子在表面横着走。而实际情况呢?原子在扩散的过程中,z方向的坐标也发生相应的变化。就如同你从山的一边爬到另一边,虽然海拔基本没变,但爬山这个过程,有升,下山过程,有降。所以,这种情况下,直接用nebmake.pl插的点在z方向上的物理意义并不准确。稍微扩展下,在x或者y方向上也可能会发生类似的情况,如果IS和FS在某一维度的变化很小时,一定要注意这一维度上的物理意义是否可以被表现出来。

既然知道了这个坑,我们该怎么填呢?
方法1:
管它是不是坑,或者不知道这个坑,直接开车冲过去。这是很多新手常见的做法,虽然有时候可以开过去,但不推荐,毕竟也会溅一车泥,搞不好坑大了还会掉进去。
方法2:
学习书中的例子,在计算之前先手动修改下结构。比如插了8个点,把第2和第7个IMAGES中的坐标向上移动0.1$\AA$。第3和6个向上移动0.15$\AA$,第4和5移动0.2$\AA$。这样做的好处就是,初始结构在z方向上具有更好的物理意义,使得计算收敛更快。
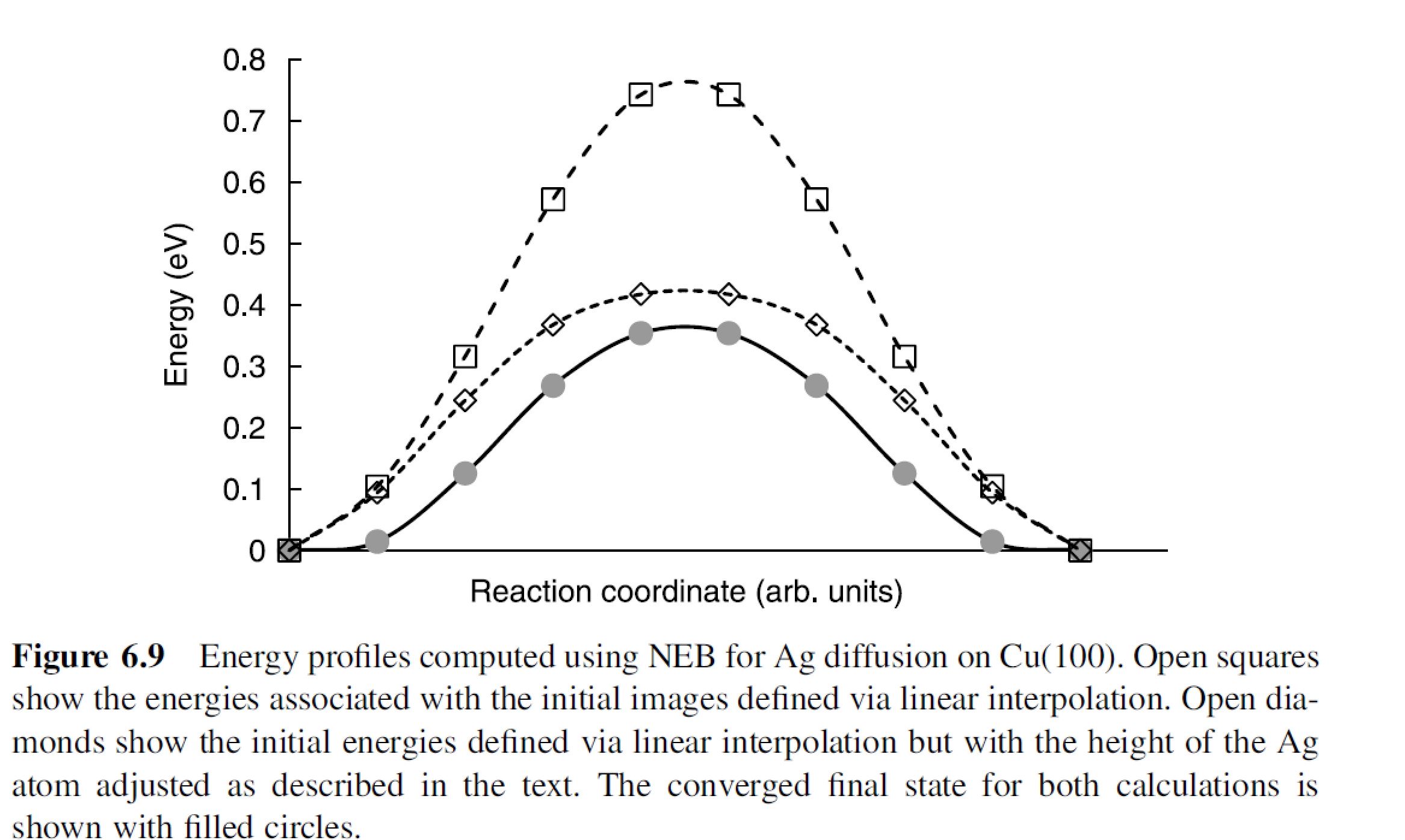
上图是书中的一个例子,最上面的曲线是没有调节z坐标时,初始结构所对应的能量;中间的为调节初始结构的z坐标后,初始结构的能量;最下面的是NEB计算完成之后,各个IMAGE的能量。从能量上也可以看出来,如果z坐标我们不修改的话,体系能量与稳定的相差甚远,间接告诉我们可能需要更多的优化步数来收敛。这个强烈建议大家自己亲手算一算。体系简单,不耗费那么多机时,有助于加深对NEB的认识和学习。