Ex69 表面吸附物种的熵
上一节我们知道怎么获取气相分子的熵以及吉布斯自由能的计算。这一节,我们简单介绍一下表面吸附分子熵的计算。
复习基础知识
首先我们应该要知道对一个体系来说,都有哪几部分对熵有贡献。一般熵通过统计热力学计算分子的配分函数来获取:主要有平动,转动,振动,电子以及核这5部分。参考下面的这个链接: http://210.45.168.34:8080/elite/wlhx/jiaocai/C_05.htm 或者找本物化书狂补统计热力学的知识。
1)电子运动的能级间隔很大,除了在几千度以上的高温条件下,电子常常处于基态。一般情况下各激发态对配分函数的贡献都可忽略。
2)原子核的能级间隔极大,在一般的物理和化学过程中,原子核总是处于基态。所以这一项也可以忽略。
3)到现在我们需要考虑的只有平动,转动和振动这三个了。
分子从气相到表面吸附的过程
当分子从气相吸附到表面上,打个不恰当的比喻,就如同图中的苍蝇被粘住在纸板上,因此就不能随便乱飞了,更不用说施展什么空中技巧了。

同样,对分子来说,举个例子,一个非线性的三原子分子,一共有3N个自由度。气相里面为:3个平动,3个转动以及3N-6个振动。但是当它吸附到表面上,由于平动和转动被限制住了,这6个自由度则会转化成振动自由度,也就是在表面上分子有3N个振动模式。如果你可视化表面上吸附分子频率计算的结果,会发现最后6个分别对应的是平动和转动,但它们已经不是气相中的平动和转动了,我们称之为:frustrated translation,frustrated rotation。

(你大妈(平动转动)已经不是十年前的大妈了,你大爷(振动)永远是你大爷)
这两个在表面化学里面是非常令人蛋疼的东西。这个我们后面慢慢讲。至少现在我们知道了,表面的吸附物种有3N个振动方式。所以,平动和转动对熵的贡献也不用管了,直接处理振动即可。(不会算的筒子们小开心下。)
通过振动频率来计算Entropy
振动频率对熵的贡献,随便查阅一本统计热力学的书,都可以找到答案,这里我们的参考书是第十版Atkins的物理化学。
Physical Chemistry 第10版的642页。获取方式:QQ群文件,Books文件下里面就有,自己下载。


图里面圈出来的公式里面,β,h,c,是基本的物理学常数。v (上面加个波浪号)是波数,单位是:m-1。我们就是通过这个公式来计算单个振动模式对熵的贡献。这个很简单,直接把数值带入公式即可,可以摁计算器,也可以用excel等其他工具进行计算。本节,我们分享一个对应这个公式的python小脚本。
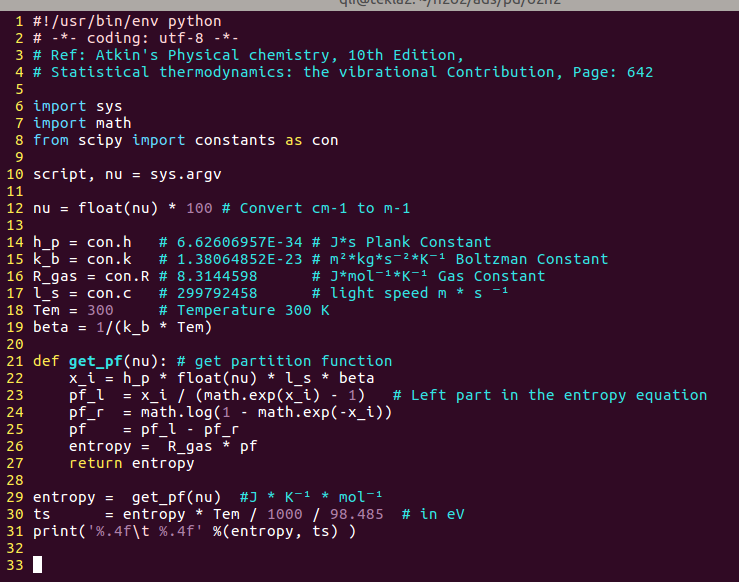
通过单个振动频率计算熵的小脚本,图里面的98.485是错的,应该是96.485,kJ/mol 转换为 eV的常数。
脚本解释:
i)VASP得到的频率波数是cm-1,这也是大家所习惯接受的,所以在12行,单位要乘以100先将 cm-1 转化成m-1。
ii) 剩下的14-19行是一些基本的物理量,我们默认温度是300K;
iii) 21-27行:是我们定义的一个计算熵的小函数。
x_i 是书里面的 βhcv
pf_l和pf_r 是花括号里面,减号左面和右面的部分
pf 得到的是花括号的结果
然后entropy 是 pf * R
iv)29-31行就是调用函数,然后输出S, TS部分了。这里S的单位是 J K-1mol-1。TS 的单位是我们熟悉的eV。
v)自己对着图片输入一遍就可以得到脚本了,不要问我要现成的。如果自己照着抄的脚本运行出现错误,反复跟图片里面的进行对比,然后进行修改。
熟悉不同的振动频率对熵的贡献
有了这个小脚本,我们就可以计算任何振动频率对熵的贡献了。一般来说,我们VASP的频率计算会有3N个波数,我们需要做的就是挨个算(虚频除外),然后求和即可。这里,大师兄想强调的并不是如何去通过这些频率去算熵(相信到现在你也已经学会了),而是要了解频率大小对熵的贡献程度。下图是我们计算了不同波数(从10到500 cm-1)的振动所对应的熵,以及TS, 单位分别为:J K-1mol-1 和 eV。
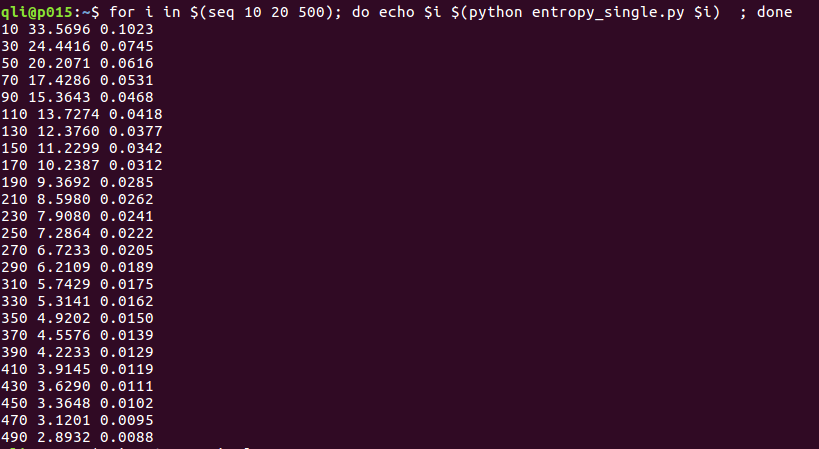
通过上图的结果,你会发现:当波数越小的时候,TS越大,随着波数增加,TS越来越小,在490 cm-1, 300K 的时候,连0.01eV都不到。一般来说,正常的振动频率(3n-6,过去以及现在的大爷那部分)对熵的贡献,基本上就可以忽略掉了,因为一般来说正常的频率波数在400-3000cm-1范围之间。
我们现在知道,波数越小,他们对熵的贡献也就越大,而前面我们说的frustrated translation 和 rotation,它们恰恰具有很小的波数,尤其是frustrated translation这部分,大部分都在50cm-1以下,因此他们对熵的贡献不能忽略。然而,它们既有体相声子谱的一些特征,也有气相分子的平动和转动的样子。因此在处理的时候,要小小心心地将它们剥离开来,非常令人蛋疼。正所谓:雄兔脚扑朔,雌兔眼迷离;双兔傍地走,安能辨我是雄雌?

另一方面:由于它们的波数很小,而VASP在小波数这部分的计算很烂,软件计算的结果也会导致很大的误差产生。一般来说大家都直接放弃,忽略这部分了。当然这样的理由很多人不服,明明知道它们波数小,对熵的贡献大,却又不得不忽略。这里,大师兄再给你另外一个解释:摘自Computational Catalysis (RSC) 这本书的第32页。如下:

在这里建议我们将平动单独处理,假设的是分子在二维平面内可以运动。也就是说只损失了一个方向的自由度。经验告诉我们,对不同的过渡金属表面来说,平动部分对熵的贡献可以作为一个常数来处理。所以,又可以不考虑了…. 以上是大家一般都不考虑这部分的贡献的原因。当然,你也可以根据上面的公式计算frustrated translation对熵的贡献,然后其他的按照振动来处理即可。
如果你对这一部分感兴趣,可以参考下这些人的工作:
1) Campbell 这个牛牛的JACS,还有一篇Chemical Review,大家自己找下。

2) 最新的一篇JPCC

3) Tamkin 这个软件的作者以及她发表的一些文章: http://molmod.github.io/tamkin/
4)Norskov 这个牛牛的一本书:Fundamental Concepts in Heterogeneous Catalysis (群文件也有,自己去找)
https://onlinelibrary.wiley.com/doi/book/10.1002/9781118892114

注意: 在这本书里的P33页,不同的吸附位点,构型数目,也会对熵有贡献:如下图:

这个的贡献不是很大,如果你的覆盖度为1/9的时候,R ln((1/9) / (8/9)) = R ln (1/8), 300K的时候,TS = 300 8.314 ln (1/8) / 1000 / 98.485 = 0.053 eV。差不多可以忽略掉。
再提零点能
师兄,既然前面的frustrated的平动和转动我们都忽略了,那么他们的零点能矫正的时候还用不用考虑。
答:小波数范围对零点能的贡献很小,一般来说,考虑或者不考虑它们的贡献,无关紧要。本人一般直接都加上。
思考
1)表面化学中,熵的贡献主要在吸附/脱附的阶段,因为分子损失/获得了大部分的平动和转动,对于表面吸附物种的熵:
i)3N-6的振动部分贡献很小,可以忽略不计;
ii)Frustrated rotation 大约在200-400cm-1左右,也可以和振动部分一起忽略;
iii)Frustrated translation部分,可以近似为二维平动来处理。也可以忽略,一般来说:忽略 是大家普遍的做法。如果理论基础不扎实,只想简单算算,那么你的腰椎间盘还是不要突出为妙。
iv)表面吸附位点的数目对熵的贡献,基本也可以忽略。
2) 而在表面反应的阶段,我们计算反应热和活化能的时候通过下面的公式:
ΔE = E(FS) – E(IS);
Ea = E(TS) – E(IS)
这两个量的计算都是通过相减来得到的,而这个相减的过程中:
i)大部分熵的贡献可以被抵消掉了;
ii)小波数范围内零点能的矫正部分也会被抵消掉,所以零点能校正的时候,大胆地直接地把非虚频的部分直接加起来就可以了。
iii)如果你不信,那么可以通过频率计算一下IS,FS,或者TS的熵,然后计算矫正过的ΔE, Ea,会发现熵的贡献很小很小,所以一般大家都直接考虑零点能,而不考虑熵的贡献,如果有审稿人问你这个问题,用脚本算一下,回答审稿人说影响不大就是了。
3)总结:表面吸附物种的熵,一路忽略,结果就是啥也不算… 不过,不算归不算,但背后的原因或者依据你得搞明白。