Ex55 简单粗暴地获取初始构型(二)
前面一节,我们通过一个简单的方法获取了一个Cu-O键的键长。因为我们固定了Cu的坐标,当计算完成之后,O原子初始构型的坐标可以直接将计算后的结果复制过来。此时需要注意Direct和Cartesian的坐标问题。
1 搭建合理的O吸附模型
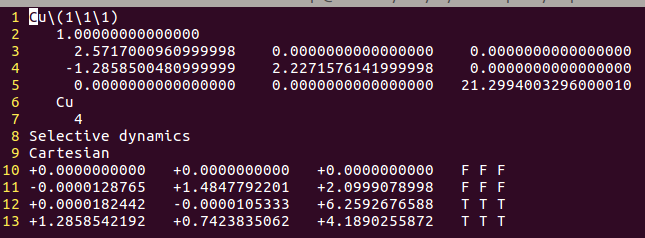
1)取Cu(111)表面模型的POSCAR (Cartesian坐标)
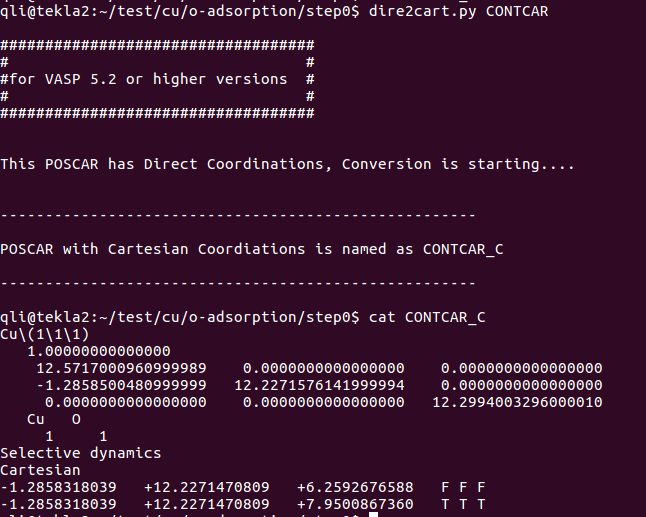
2) 复制快速获取初始构型计算(Cu-O dimer)的结果
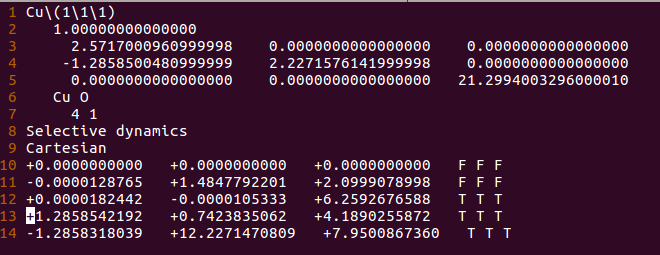
3) 构建O的吸附模型:
当然了,我们也可以根据键长或者O在z方向的坐标直接在Cu(111)表面基础上搭建。
这个简单粗暴的方法到此就讲解完了。有以下几个需要注意的地方:
A)这里我们用的是Cu(111)的 p(1x1) slab模型,表面只有一个原子。一般来说,大家计算吸附的时候,模型都比这个大,我们可以取一层原子。记住,要固定住这层原子。
B)这个方法基于的是气相中的计算,因此偶极矫正不要加!因为加上去之后,收敛会变得很困难。这个大家可以用自己测试测试。
下面我们讲一个基于slab模型的方法,虽然比这个计算量大,但也异常的快,相对于后面的计算,花费的时间也可以忽略不计。
2 快速获取初始构型方法(二)
1)在这个方法中,第一步我们先取slab模型的结构,这里就不讲了。
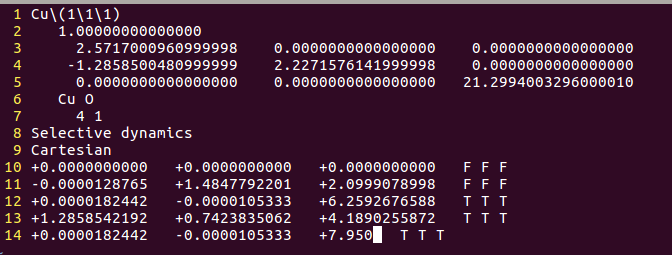
2) 搭建一个初始的吸附模型。此时我们要根据原子半径,大体确认一下键长的合理范围,前面也讲过了,就不再细说了。 由于前面一节我们已经有了一个O原子的坐标数据,可以直接拿过来。如下:
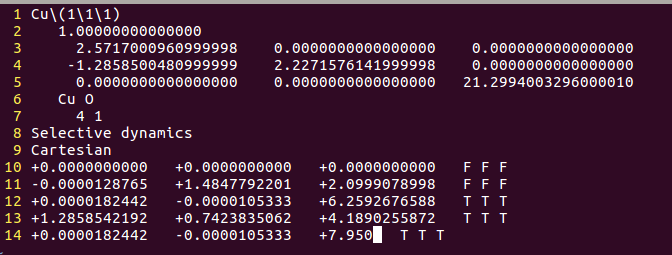
注意:实际操作的时候,这个方法可以使用前面一节的方法的出来的结果;也可以直接设置初始值,这两个方法之间联系不太大。
3) 固定slab模型中的所有原子!所有原子!所有原子!
重要的事情说三遍,是把slab模型中的原子全部固定!只放开吸附的分子。这里大师兄在vim里面操作,使用了下面的命令::12,13s/T/F/g 意思是把12和13行中的T全部替换成F。当然也可以使用sed进行操作,相信大家经过这么长时间的练习,已经掌握基本的Vim和sed操作,就不浪费时间了。效果如下:
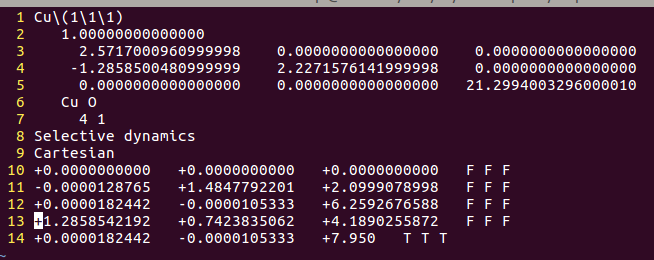
4) 设置INCAR,这里是slab模型,把之前Cu(111)表面优化计算的INCAR直接拿过来了。
EDIFF、EDIFFG 可以适当放宽,下图大师兄懒得改了。
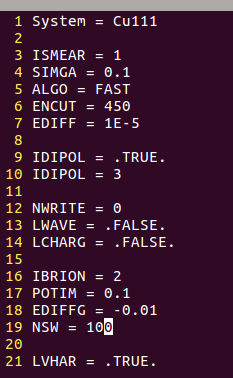
5) 生成对应的POTCAR,K点只用gamma点!K点只用gamma点!K点只用gamma点!
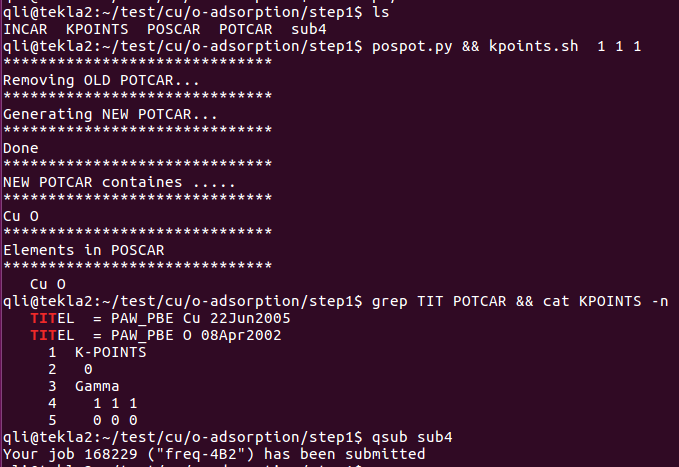
这里需要注意的有两点:
A) 计算前一定要检查POTCAR和POSCAR是否对应,养成这个好习惯
B) 一定要用gamma点。因为我们知道,K点越多,计算量越大。
6) 提交任务等待结束。
O原子的坐标,从最初的7.950优化到了8.009。
到此,另外一种快速获取稳定构型的方法,也介绍完了。希望大家可以举一反三,运用到自己实际的课题计算中。
3 正式优化O的吸附
前面我们已经获取的初始的一个合理的构型,下面就开始正常计算了。想一想,在上一步的基础上,正式计算的时候,我们有哪几个需要修改的部分。
1) 结构
这个是必须要想到的。怎么做呢? 直接将CONTCAR 复制成 POSCAR即可。
PS: 怎么完成复制这一步还不会的话,请从头开始学。
2) 复制完了并不代表完事,前面一步我们还固定了表层的原子,此时我们需要放开。使用sed命令:
1 | sed -i '12,13s/F/T/g' POSCAR |
3)KPOINTS:使用前面Cu slab优化时用的 13x13x1
4) INCAR中,如果前面一步的计算精度较低,这时,我们就要提高一下了。
5)POTCAR 不变
6)提交任务的脚本:由于前面的计算量很小,一般可以使用小队列进行计算,如果计算量增大了,这时我们也需要稍微修改下脚本。
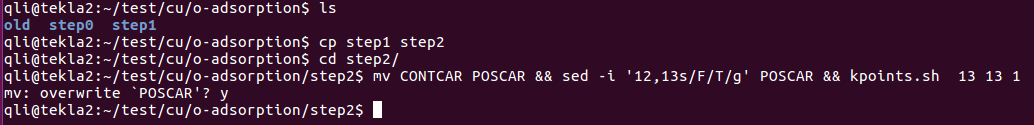
7) 提交任务,等待结束,查看结果。
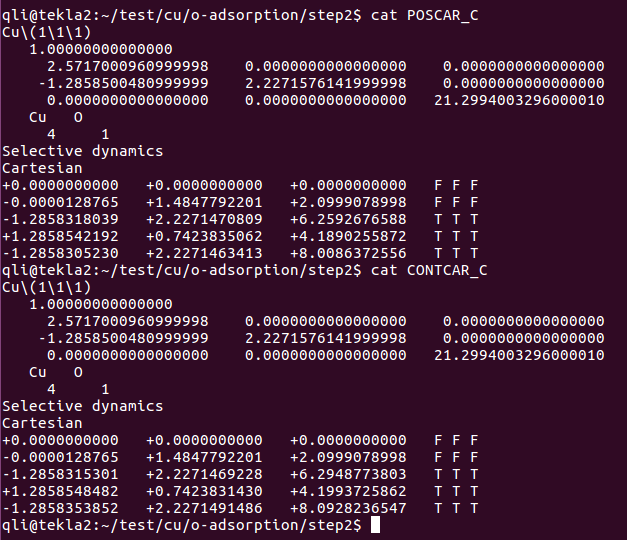
O原子的坐标从8.009 优化到了8.093。键长从1.75 Å 优化到 1.80 Å,变化很小,这说明我们的初始构型已经很接近优化结果了。
4 扩展练习:
1) 本节的实例中,使用了 && 这个连接前后的命令,大家学习下是怎么回事
2) 重复本节练习,掌握其中的关键点。
5 小结
一个合理的初始构型可以极大地降低我们的工作量,这一点是大家务必要记在心里的。而获取合理的初始构型,一方面需要我们用化学的知识去搭建结构,另一方面,也需要配合一些简单的低精度的计算去完成。希望大家可以掌握这两节的精髓,运用到自己的课题中,提高自己的计算效率。