Ex54 简单粗暴地获取初始构型(一)
$\require{mediawiki-texvc}$
Ex54-Ex56主要介绍一下如何计算:单个氧原子在p(1x1)-Cu(111)表面top位上的吸附。在实际的计算过程中,一个好的初始结构会极大地加快并节约你的计算时间,这不仅仅体现在服务器运行的时间上,也会避免很多错误的结果,因为这些错误结果的纠正耗时费力,通常折磨得新手们心力交瘁。可以回顾下前面我们O$_2$ 分子的优化过程:不合理的初始结构导致的错误结果以及计算时间的增加。本节主要介绍一个快速获取优化初始构型的方法。该方法简单粗暴,可以让你在极短的时间内快速获得一个化学的feeling。
1 Top site 的构型
在Cu(111)表面上,top位指的是直接吸附在Cu原子上方。其他的位点是什么,先不要着急,后面会慢慢学。如果一个O原子吸附在Cu原子上方,在空间坐标上,大家很容易想到,O和Cu在xy方向的坐标应该是差不多的,z方向O的坐标为:Cu的z坐标 + Cu-O键的键长。如下图:
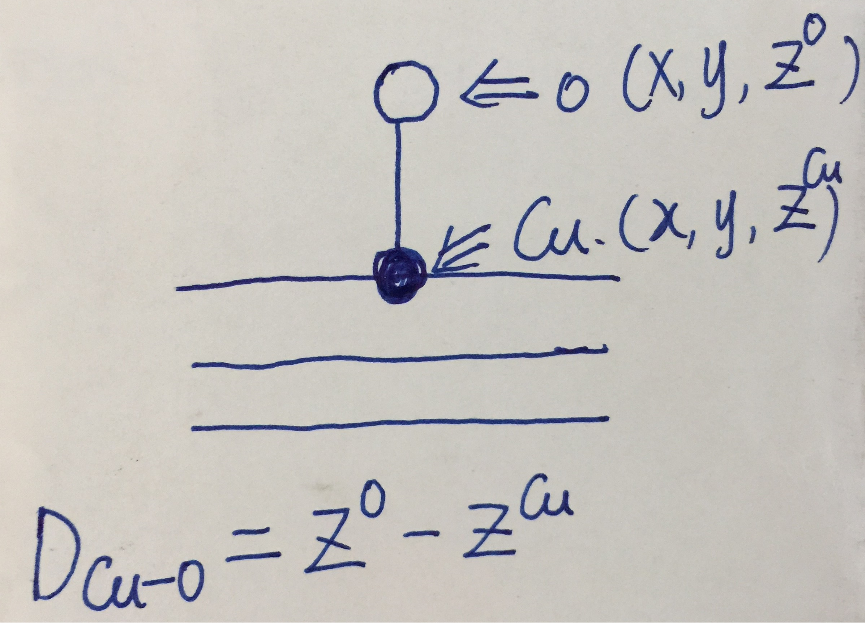
2 确定O原子在z方向上的坐标
知道了前面的基本原理,O原子的坐标我们就可以通过Cu-O的键长来初步获得了。那么Cu-O的键长怎么获得呢?这里列举了一下几个常用的方法。
1)查数据库:
2)查文献:自己去查,不要在QQ群里让别人给你发文献。
这两条主要考验的是大家查询资料的能力,这里暂且不详细介绍了。有兴趣的可以加入大师兄文献互助超级群,跟众多文献检索大牛学习。(群号:157099073)
3)自己估算
估算的话,可以根据Cu和O原子的半径:Cu和O成键,键长肯定要小于两者的半径之和。搜一下维基百科,Cu的半径为1.35 Å,O的为0.60 Å。(维基百科,搜索Atomic Radius就可以得到下面这个表)
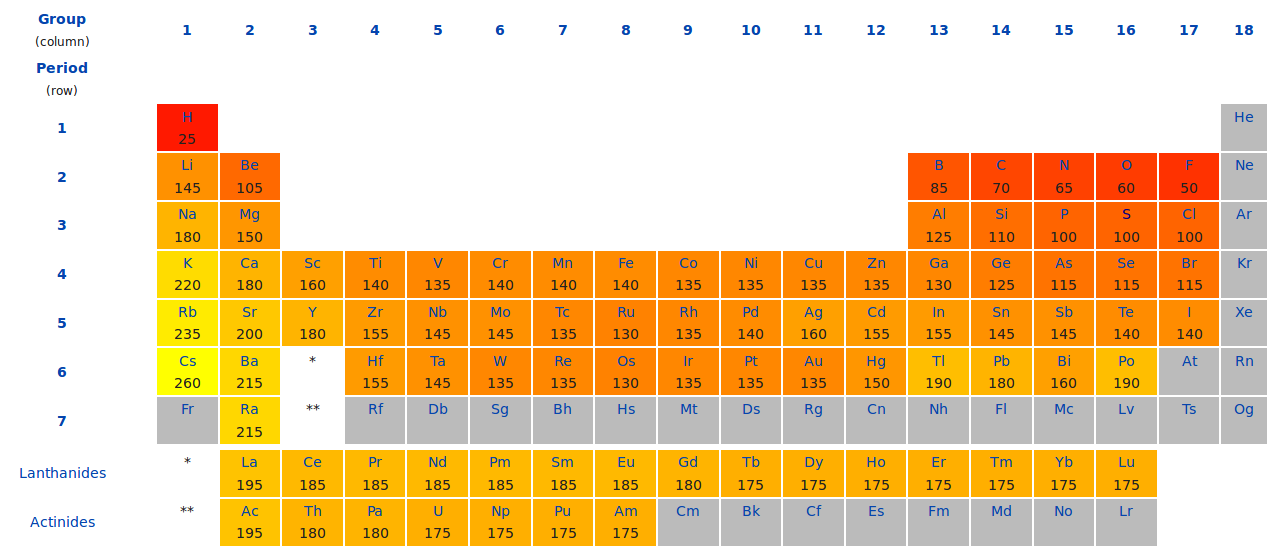
从上面的数据我们可以得出:Cu-O键要小于1.95 Å。在吸附构型搭建的时候设置比1.95 Å小点的值作为初始,进行优化。一般来说,原子半径之和减去0.1-0.3 Å都是可以的。但是不能太小,否则第一步优化的时候排斥力太大,会导致计算出错。
4)自己初算:
初算就是初步采用一个小的模型,简单优化一下,得到一个合理的键长数值。小的模型主要指2个方面:
结构简单;
计算参数简单。
下面我们主要在估算的基础上,介绍一个初算的方法:直接优化一下气相中Cu-O双原子分子的结构。
这个结构不难搭建,将前面练习中O$_2$分子中的一个O原子换成Cu进行优化即可。
为了加深大家对搭建模型的印象,我们从p(1x1)-Cu(111)表面的结构出发,然后一步一步搭建CuO的气相结构模型,并计算。
3 搭建初算的模型(Cu-O双原子结构)
1)p(1x1)-Cu(111)表面的结构
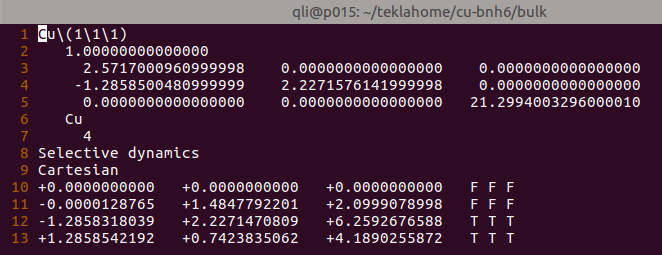
将前面几节计算的一个真空层为15 $\AA$的例子直接拿过来。表层原子在z方向的坐标为6.259 (第十二行)。
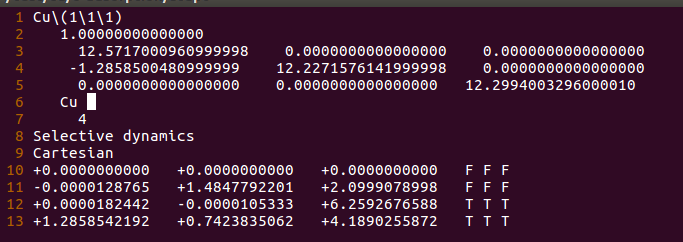
2)修改格子大小(3-5行)
这里修改的很随意,第三行中直接把2.571改成了12.571;
第四行中把2.227改成了12.227。
第五行中将21.299改成了12.299
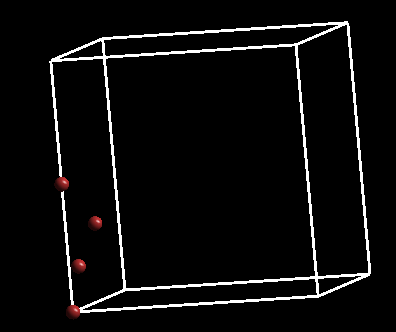
效果如下:
注意:
A)当然也可以用其他大小的格子;(例如:8x8x8 $\AA^3$)
B)格子大小直接影响计算量和时间。(回顾下前面所学)
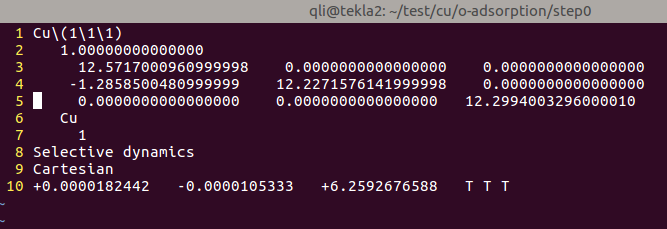
3)保留表层的Cu原子,删除其他的Cu原子。
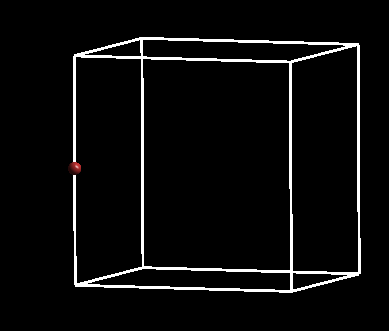
注意:
A)我们这里将表层以下的三个原子删掉了,只保留z坐标为6.259的这个原子;
B) 第7行中原子数目也要相应的改变。从4 改成1。
C) 效果如下图:
4)加入O原子
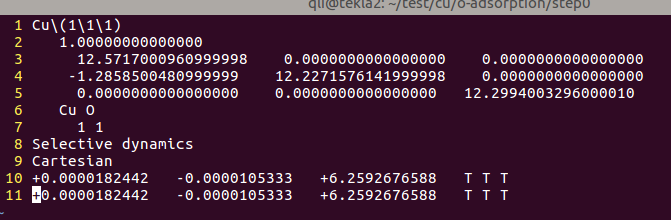
注意:
A)第6行中,在Cu的后面加上了一个O,不是数字0;
B)Cu 和 O 之间用1个或者几个空格隔开,不要用Tab;
C)第7行中,记得加上O原子的数目
D)最后一行添加O原子的坐标,这里我们直接把Cu原子的复制过来了。

由于两个原子坐标一样,Cu和O堆在一起了。
5)修改O原子坐标
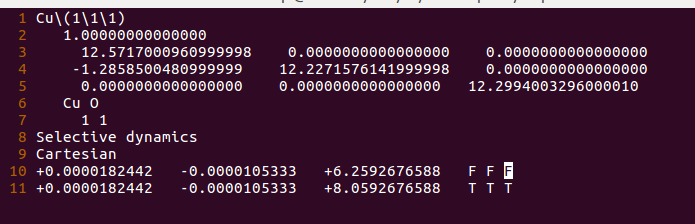
注意:
A)这里我们把Cu固定住了;为了方便下一步的计算。
B)Cu-O之间的键长,设置的为1.8 Å;
C)O的z坐标为:6.259 + 1.8 = 8.059。
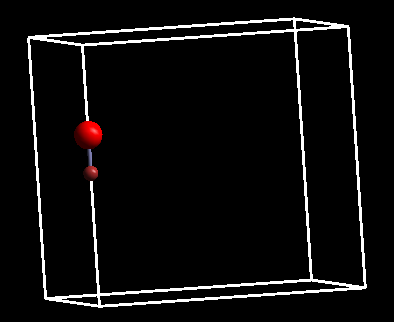
6)到这里,Cu-O双原子分子的气相结构就搭建完毕了,保存成POSCAR即可。
上面的效果图是本人每一步打开展示给大家的。实际操作中,完全没有必要。我们需要学习的是:
A)怎么将脑子中的模型转化为VASP的POSCAR文件;
B)格子大小怎么修改;
C)怎么添加原子,添加或者删除原子后,原子数目怎么弄;
D)怎么添加原子坐标。
E)怎么通过改坐标修改原子位置。
4 INCAR检查
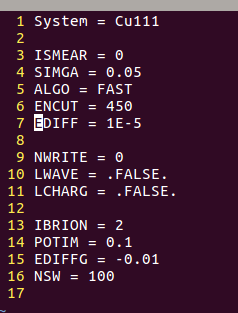
注意:
1)算的是气相中的:ISMEAR = 0;SIGMA = 0.05 (本书前面就讲过了)
2)IBRION = 2;POTIM = 0.1;NSW =100 是优化的参数
3)EDIFF和EDIFFG是电子步和离子步的收敛标准。
师兄,磁性呢?对称性呢?
因为本例子是一个初算,几步算完,看下键长。这个任务的使命就完成了。
很多细节的东西可以暂时不用考虑。这里EDIFFG用的也有点小,本人忘了修改了。大家可以设置为 -0.05或者直接使用能量作为标准,这些都可以的。
虽然本节我们很多地方都没有考虑,这是由于任务的性质所决定的。我们对其定位就是瞎算下,得到一个初始的构型。这也是一个课题中为数不多的,可以为所欲为的计算了。但你正儿八经开始算的时候,各种细节问题都要考虑进去。而这一步的计算,也可以作为一个缓冲期,让你有充足的时间去思考一下正式计算时其他需要的注意事项。
5 POTCAR、KPOINTS检查
A) 根据POSCAR获取对应的POTCAR:见附录自动生成POTCAR的脚本。
B) 使用gamma点计算。cat POTCAR
注意:
在提交一个任务前,一定要将INCAR、KPOINTS、POSCAR、POTCAR以及脚本在心里默念一遍。然后对应的检查一下,是否有些遗漏的地方。否则等算完了,发现错了,又得浪费很多时间重新计算了。
6 检查结果

1)上个厕所的功夫,任务就结束了,共算了6步。(其实是切换电脑系统的功夫)
2)Cu—O 的键长:7.95-6.26 = 1.69 Å
注意:
此时的键长只作为下一步的初始猜测,如果和文献去比有点差异,也不要太较真。
7 总结:
本节,你会学到:
1)如何通过原子半径,估算一个初始的键长值。
2)如果搭建一个简单的模型,初算一个键长值。
3)复习气相中的优化。
4)复习下计算前准备工作:INCAR、POSCAR、POTCAR、KPONTS的检查。
如果本节的内容,你有不理解的地方;想获取脚本等信息;请查阅前面讲的以及附录中的内容。搭建几何模型,无非就是按照软件的格式,修改原子坐标罢了。这里我们强调的是几何模型。但,几何模型所具有或者要表达的物理、化学意义,这才是最关键的。