Ex23 乙醇分子的振动频率计算(一)
大师兄在本节开头放上这张图片的意思是,请大家再仔细体会Rubbish in, Rubbish out这句话,只有对我们要研究的体系有一定的理解,明白我们具体要计算什么内容,设置好输入文件的参数,才能得到我们需要的合理结果。接下来的几节会涉及到分子的振动频率计算,振动频率的可视化等内容,请大家跟着大师兄一起练习,掌握振动频率的基本计算和分析方法。
1 分子的振动
我们首先看回顾一下振动相关的基本知识,这里大师兄不具体解释,引用 $9$ 版 $Atkins$ 的《物理化学》书中的内容,书已上传至QQ群文件中,也可百度网盘下载: http://pan.baidu.com/s/1o8HlyOi 全是英文,大家耐心阅读下。
简谐振子、胡克定律、体系势能随着振动距离 $x$ 的关系

简谐振动的薛定谔方程描述:

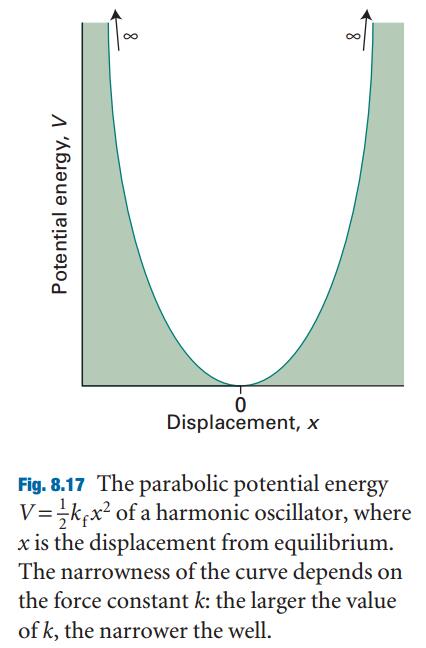
薛定谔方程的解:振动的量子化,振动频率

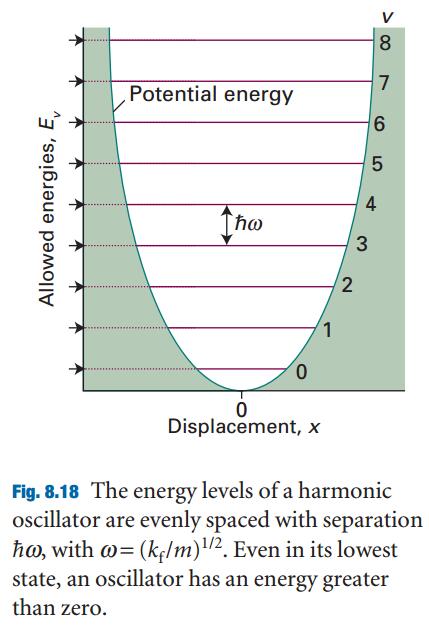
零点能的数学和物理两个方面的解释:

2 频率计算的作用
频率计算有什么用?为什么要算频率? 大师兄稍微总结了一下频率计算的意义,大体有以下几个方面,没有提到的用处,烦请大家指出来,以便补充。
2.1 确定结构是否稳定;
2.2 看振动方式和大小,用来和实验对比,棋博士最新的文章就是一个非常好的例子;
2.3 反应热,反应能垒,吸附能等的零点能矫正;
2.4 确认过渡态(有一个振动的虚频)
2.5 热力学中计算entropy,用于计算化学势,微观动力学中的指前因子和反应能垒。
3 怎么用VASP计算频率?
3.1首先进行结构优化,获取稳定的构型,这个我们前面已经讲过了;
3.2 将原来的CONTCAR复制成POSCAR :1
cp CONTCAR POSCAR
3.3 修改INCAR
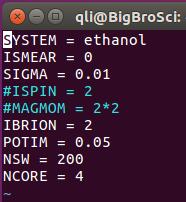
修改后如下:
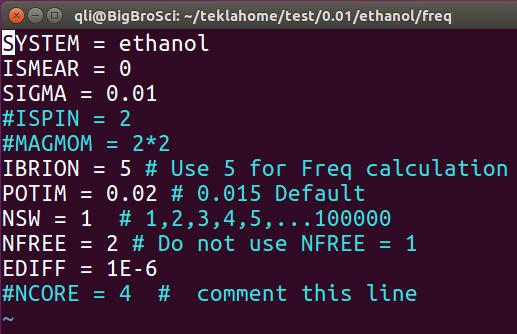
频率计算的INCAR
IBRION的值改成5POTIM用一个更小的值,我们这里用的0.02,默认值是0.015NSW设置成1,这个可以直接不管,继续采用优化时的NSW值,因为你设置成1, 2, 3, 4, 5, …, 1000都不会影响计算;但不能不设置(因为默认值是0,这时算个单点后任务便停止了。)NFREE=2添加这一个参数,表明原子在某一方向上正反两个方向移动;NCORE=4这一项要注释掉!大师兄这边的服务器,并行计算频率时 VASP 会罢工,只进行一步静态计算,注释掉就正常进行了;此外,
EDIFF也要设置一个严格的值(频率计算时,默认值为1E-6,足够了!下一节会讲到)
小结一下频率分析关键的参数:
1
2
3
IBRION=5
NFREE=2
POTIM=0.02
1 | IBRION=5 |
4 扩展练习
4.1 按照本节的流程新建一个文件夹 freq : 该文件夹中包含乙醇分子优化后的结构 (将CONTCAR复制成POSCAR),以及优化时的POTCAR,INCAR, KPOINTS以及提交命令的脚本文件;
4.2 修改乙醇分子优化的INCAR为频率计算的INCAR;需要修改哪些参数心里要清楚;
4.3 运行乙醇分子频率计算,并查看频率分析的OUTCAR,OSZICAR等输出文件;
4.4 查看 VASP 官网对于IBRION=5 的解释,搜索网上相关频率计算的文章,帖子,初步了解NFREE,POTIM所代表的含义;
https://cms.mpi.univie.ac.at/wiki/index.php/IBRION
4.5 查找官网中频率计算的例子:
A) https://cms.mpi.univie.ac.at/wiki/index.php/CO_vibration
B) https://cms.mpi.univie.ac.at/wiki/index.php/H2O_vibration
5 总结:
5.1 熟悉频率计算初始文件的准备过程;
5.2 频率计算INCAR中的三个重要参数;
1 | IBRION = 5 |
5.3 初步了解频率计算中各个参数的含义。