Ex22 乙醇气相分子的优化
前面我们终于讲完了 $\rm{O_2}$ 分子优化的例子。相信大家对 VASP 计算已经有了一个初步的理解。这一节我们继续学习气相分子的优化。为了让大家进一步了解计算的过程,我们选取一个稍微复杂的分子作为例子:乙醇($\rm{CH_3CH_2OH}$)。
问:把大象装进冰箱需要几步?

大师兄,这个问题我早就知道答案了。为啥还问这样低智商的问题?
大师兄要求的不是让你回答开冰箱门,装进去,关门的这三步。而是让你尝试回想一下:当你第一次接触这个问题的时候,你的反应是什么?
大师兄比较笨,我的第一反应是,这怎么可能? 冰箱那么点,大象那么大。

当朋友告诉我答案的时候,才恍然大悟,这跟大小没关系。
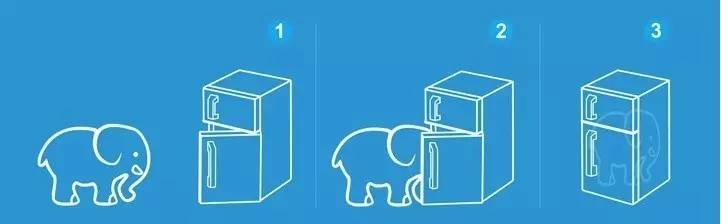
同样的,怎么用 VASP 计算乙醇分子?很多童鞋就如同第一次被问到大象这个问题一样不知所措。答,也是三步!
1) 打开冰箱:准备 VASP 文件
2) 把大象塞进去:准备乙醇分子模型
3)关上冰箱:运行 VASP
1. 打开冰箱:
我们可以直接用O$_2$分子计算的输入文件,
1.1 复习一下前面学到的INCAR和KPOINTS的内容:
1)乙醇分子是闭壳层的分子,没有磁性,不需要ISPIN=2
2)气相分子计算,我们要用ISMEAR=0,SIGMA取值要小,SIGMA=0.02;
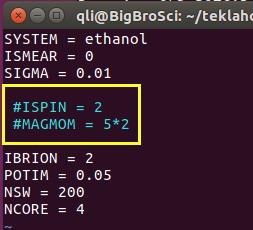
# 表示注释,这个符号后面的内容,VASP在运行的时候不考虑。
示例 1 :1
2ENCUT = 400
# ENCUT = 500
和1
ENCUT = 400
效果是一样的。
示例 2:# 的用法:
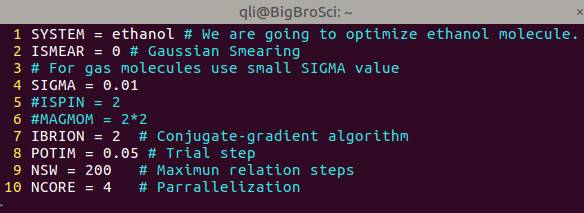
VASP 计算中一些常见的错误,以及注意事项,你可以通过 # 写在INCAR里面,方便计算的时候进行设置。可以 # 开头,单起一行,也可以在参数的最后面加上注释。新手们刚刚开始,可以结合 VASP 官网参考书,把用到的INCAR参数注释下来,时间长了慢慢就掌握了。
3)气相分子计算,K点使用Gamma点就够了;
1 | K-POINTS |
2 把大象塞到冰箱里面 :POSCAR的准备(本节重点)
前面我们学习到,一个好的初始结构会加快计算,获取准确的计算结果。因此我们需要去找一个合理的模型。对于气相分子的结构,一是手动搭建,大师兄推荐用 GaussView,另一个办法就是找数据库,大师兄推荐英国皇家学会的 ChemScpider。网址:http://www.chemspider.com

下面我们把手动搭建乙醇分子的模型具体解释一下:
2.1 打开网站
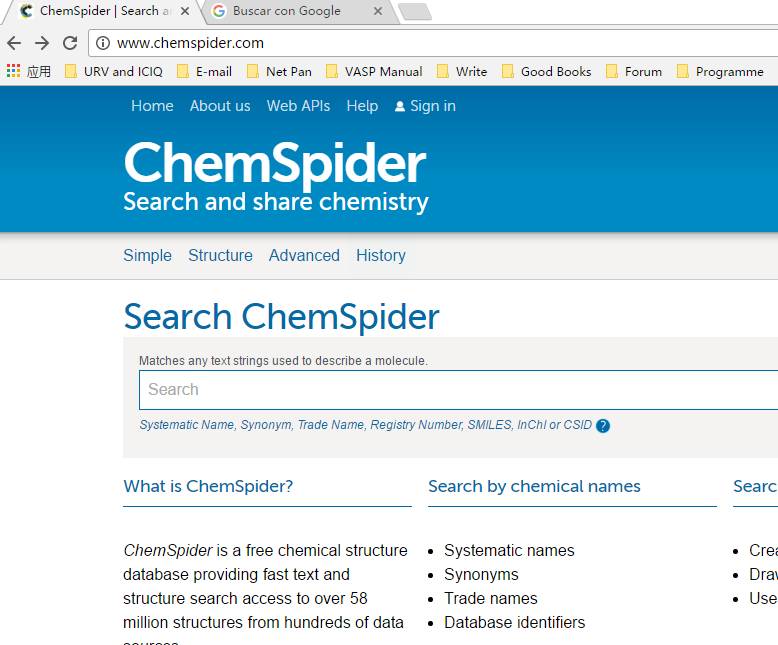
搜索框中输入: 分子名称Ethanol 或者 分子式C2H6O…. 点击搜索,等待结果:
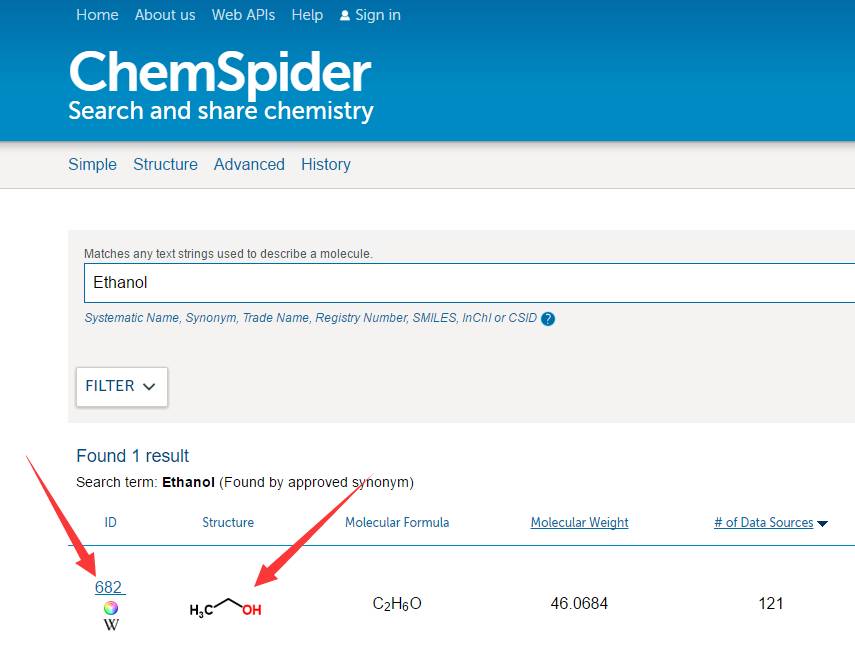
上面箭头指的两个地方随便点,效果是一样的,如下:
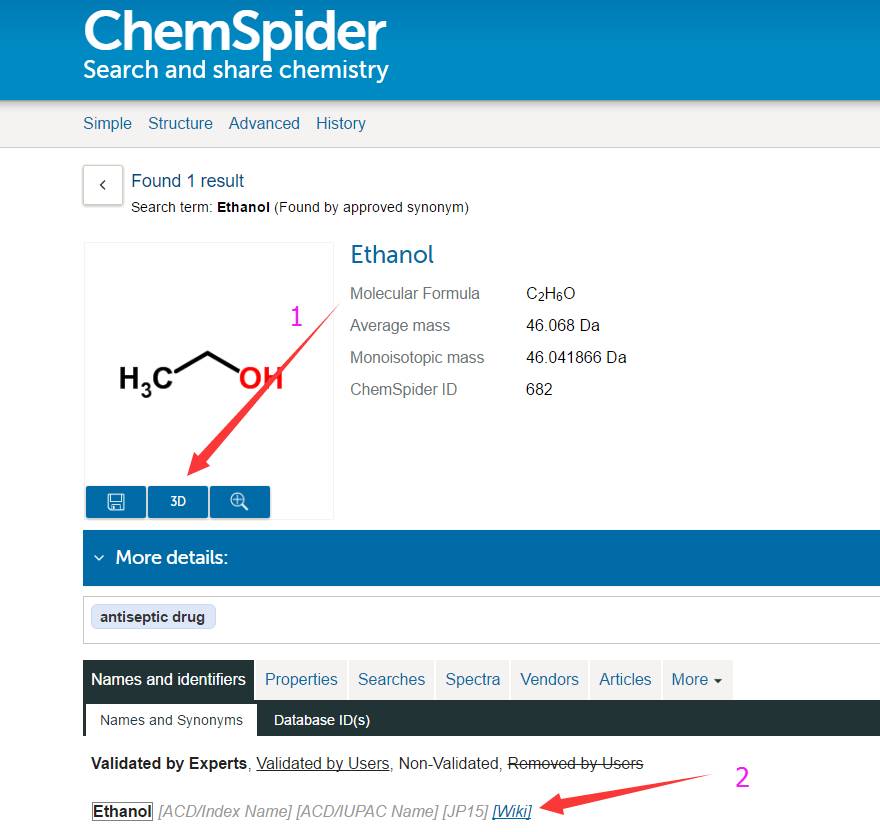
分子默认显示 2D 的结构,点击箭头指的 3D,切换。第二个箭头所指的有对应结构的维基百科链接,翻墙的筒子们可以查阅下相关结构的知识。选择3D后,如下:
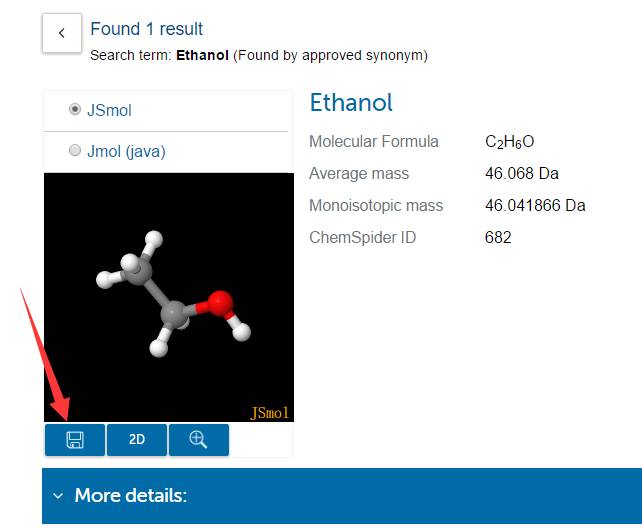
点击箭头指的地方保存,浏览器会下载对应的 .mol 文件,文件名为该结构在数据库中的编号。
.mol文件也是纯文本,使用Notepad++ 打开如下:看到这么多内容,不要害怕!
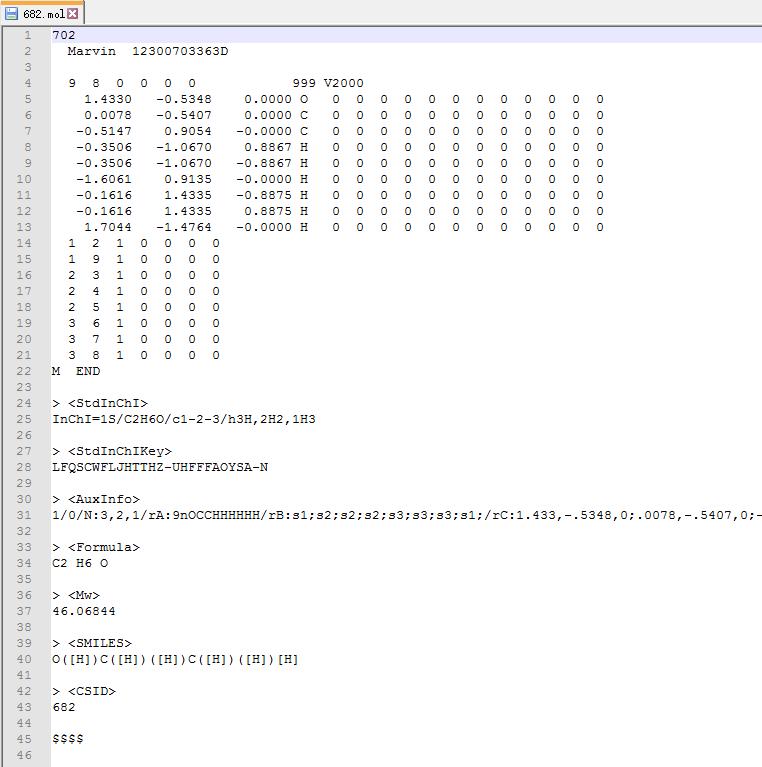
注: 不仅仅是 .mol 文件,很多结构文件都是文本格式,直接打开就是。
从里面找到乙醇结构的 $x, y, z$ 坐标信息
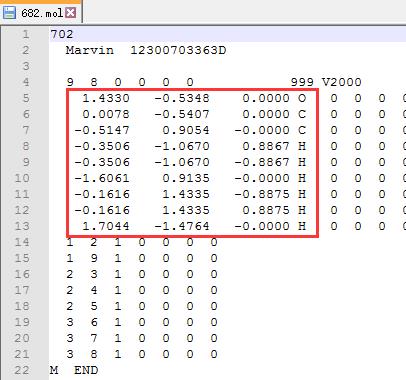
删除上图中除红色框之外的所有行,1-4行,以及 14 至最后一行,只保留 $x, y, z$ 坐标信息的那几行。
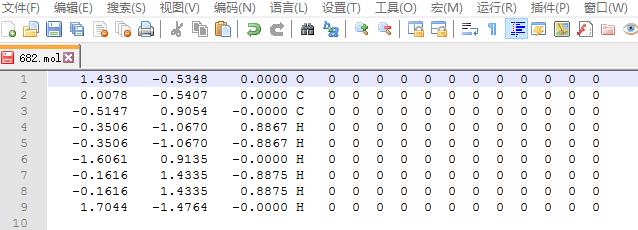
手动写POSCAR,首先你要熟记的格式,知道POSCAR从头开始,往下每一行代表的内容。
我们的模型是把乙醇分子放到一个 $ 20 \times 20 \times 20~A^3 $ 的格子里面。输入完之后,坐标后面的元素符号以及那些 $0$ $0$ $0$ 可以删掉,也可以不管。
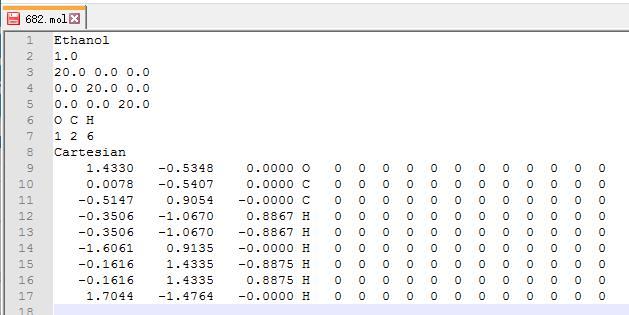
关键点1: .mol文件中,坐标那几行中的第 4 列写到POSCAR中的第 6, 7行!
关键点2: 注意坐标为 Cartesian 或者是 Direct. Direct 是分数坐标,其 $x, y, z$ 值都小于 1.
如果你想删掉图中坐标第3列后面的内容:大师兄推荐notepad++里面的列块模式:如下:
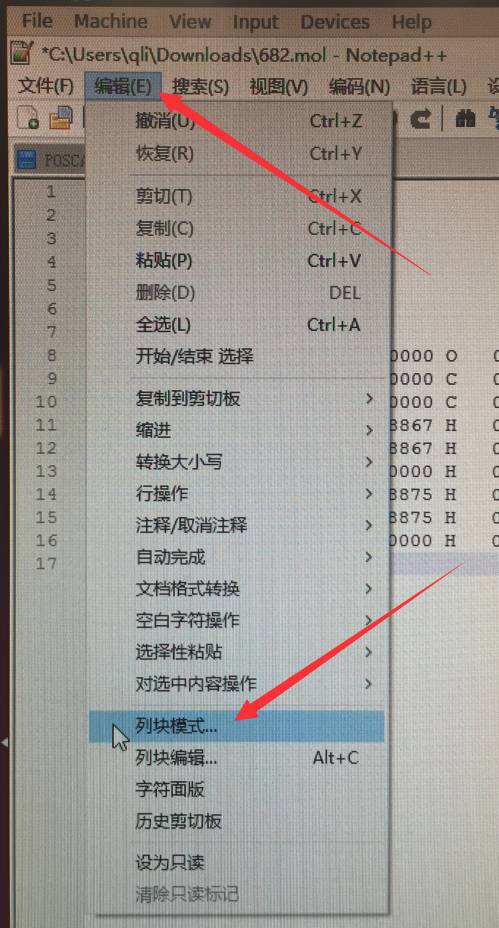
点击之后,可能会弹出对话框,告诉你如何使用,列块模式
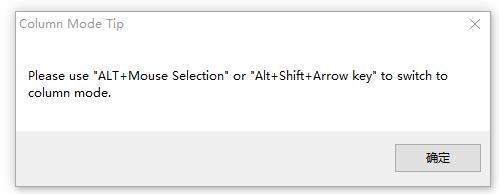
方法1:
摁住 Alt 键,然后用鼠标选择文本,不同电脑可能不一样,大师兄这边同时摁住 Ctrl 和 Alt 两个键,然后用鼠标选择的。
方法2:
同时摁住 Alt和Shift键,通过键盘上前后左右的箭头选择文本
大家可以尝试下,选中效果如图:
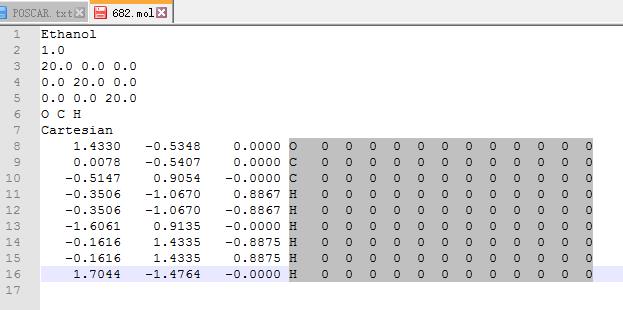
然后点键盘上的Delete键删除.
然后另存为POSCAR即可。
我们可以使用 p4vasp 来查看一下模型的结构:如下图
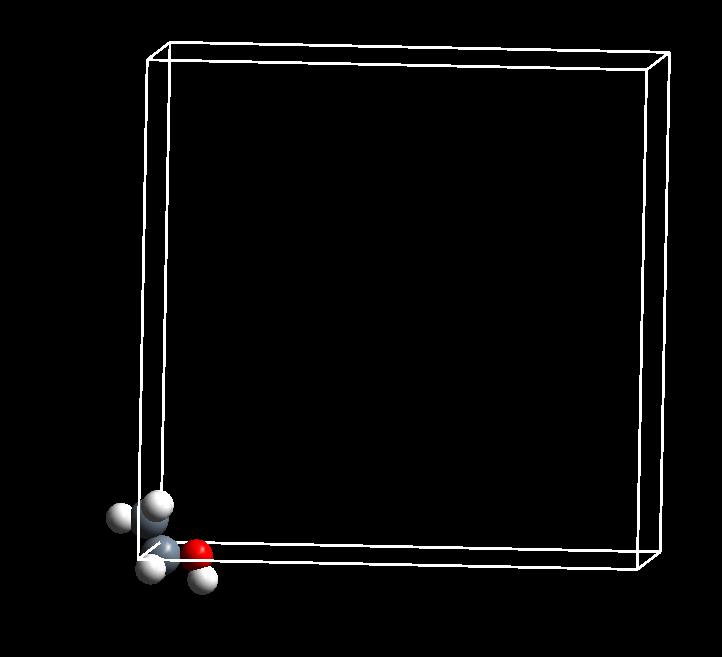
我靠,师兄,结构怎么跑到格子外面啦?前面 $\rm{O_2}$ 分子的学习中,你已经知道了这是因为周期性导致的显示问题。对计算不会产生影响。这个结构可以拿来直接用。
如果感觉不爽,想把结构放到中间,可以这么做:
数学上,把 $x, y, z$ 坐标统统加上 $10$ 即可;
软件使用上,我们讲一下 p4vasp 的操作方法:
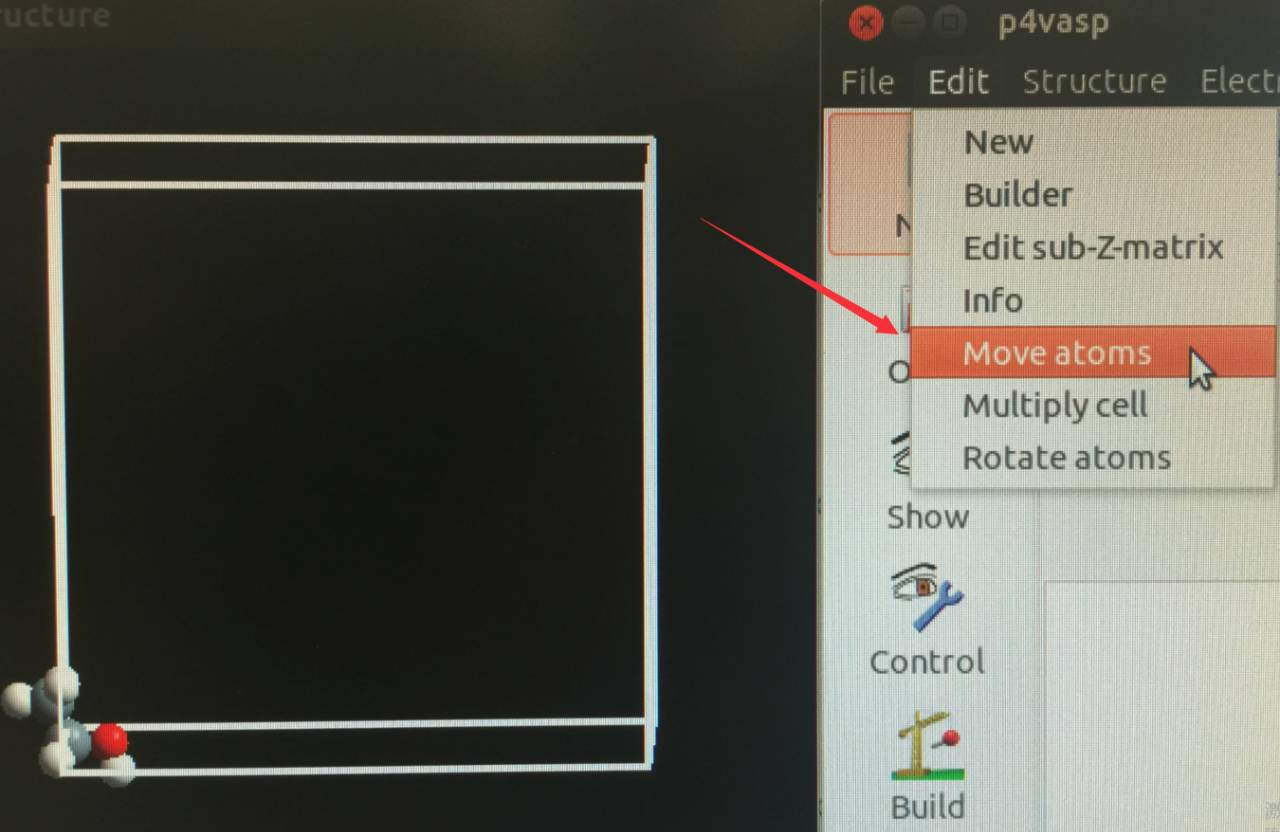
选择 edit –> Move atoms
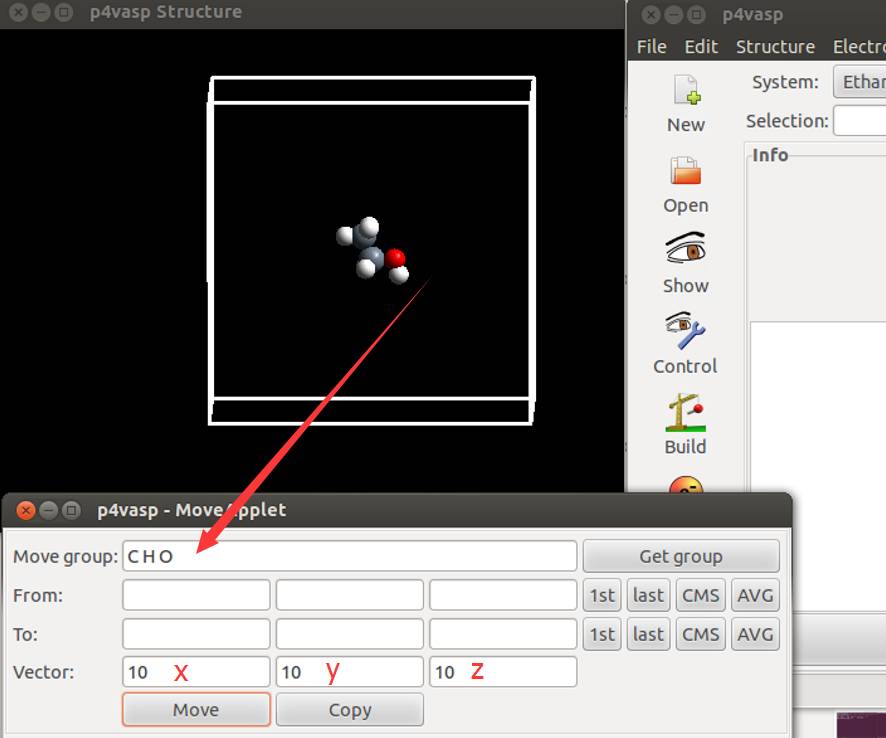
Move group 是你要移动的原子,这里大师兄直接输入了 C H O 三个元素符号(中间有空格)表示选择所有元素的原子。然后在 Vector 中选择 $x, y, z$ 三个方向上移动的大小。你也可以写 $1$ $1$ $1$,然后点击 Move 按钮 $10$ 次。
如果你想通过选择原子来实现移动的话(不直接在 Move group 里面输入 C H O),需要按照大师兄说的步骤走:
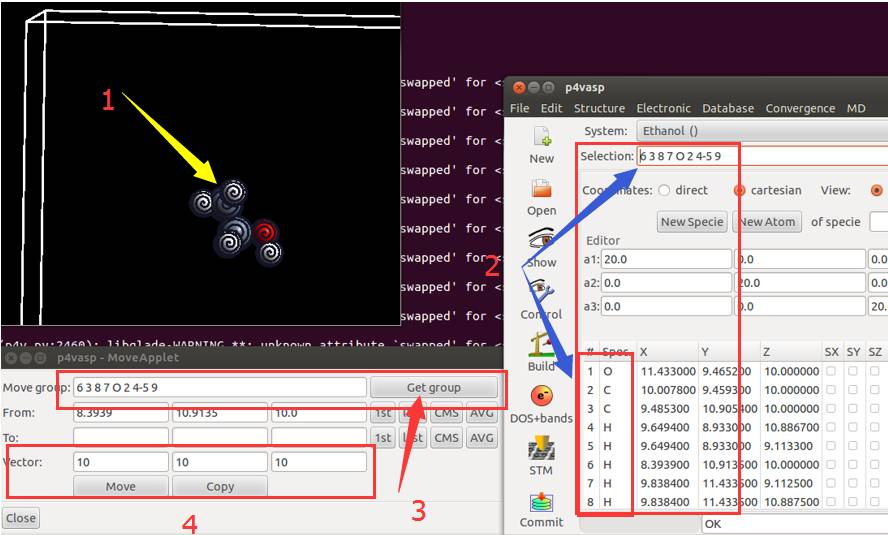
1) 空格键结合鼠标选中所有的原子;鼠标指到原子上就点一下空格键
2) 选中原子后,主界面会显示一些数字,这些数字和 POSCAR 中元素的顺序是一致的;
3) 所有原子选中后,左下角的框中点击 Get group,会显示选择的那些原子;
4) 在 Vector 中选择 $x, y, z$ 三个方向上移动的大小,然后点击 Move 按钮。
上面一堆废话就此打住,结果就是这样子的:
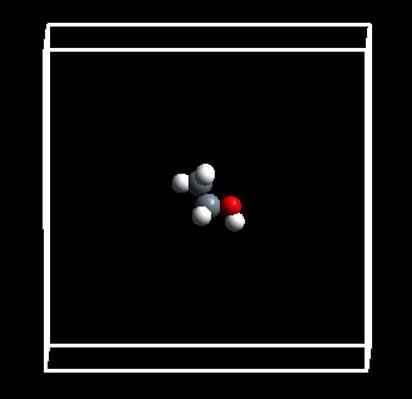
然后点击 file -> Save system as -> 选择目录 -> 保存成POSCAR
POTCAR
POSCAR 讲完了,我们就要按照里面的元素顺序制备POTCAR了。
首先: 我们要准备 O C H 三个元素的POTCAR,去POTCAR的数据库中去找:
然后复制到当前目录下,三个元素的POTCAR分别命名为:POTCAR-O, POTCAR-C和 POTCAR-H, 把三个元素的POTCAR合并在一起,命令就是
1 | cat POTCAR-O POTCAR-C POTCAR-H >> POTCAR |
3 提交任务
提交任务之前,需要再次检查自己的输入文件一遍,没有问题,提交直至结束。
4 扩展练习:
4.1 从头到尾,认真重复本节中大师兄的操作;
4.2 记住本节讲解的内容,自己重复一遍操作,直至自己通过文本编辑器会搭建结构模型;
4.3 运行乙醇计算的例子;
4.4 尝试使用 GaussView,Materials Studio (MS),以及其他可视化界面搭建模型;
4.5 学会使用 VESTA 导出POTCAR格式的结构数据。
5 总结:
本节,我们主要讨论了一下分子模型的数据库搜索和搭建工作。希望大家能够完全掌握本节的所有内容和细节。在计算中,你会遇到各种各样的结构文件,其实都是$xyz$坐标的衍生物而已。不要害怕,直接打开它们,学会提取里面有价值的信息。