Ex11 O$_2$单点计算和优化结果分析
前面几节,我们讲解了O分子单点计算和O$_2$的POSCAR和POTCAR的准备。这一节我们主要讲解一下:
1)O$_2$的分子结构分析
2)如何初步进行构型优化计算。
O$_2$ 的单点计算
首先解释下什么是单点计算:顾名思义就是不优化结构,直接算个能量,电子相关的性质。你也可能会听到很多人说静态计算,或者自冾计算。其实都一样的:几何结构计算前后不发生变化。
首先提交O$_2$ 静态计算任务,运行,等待结束。如果你的任务出错,请跟大师兄的输入文件进行对比、改正,直至计算正常结束。如下图:
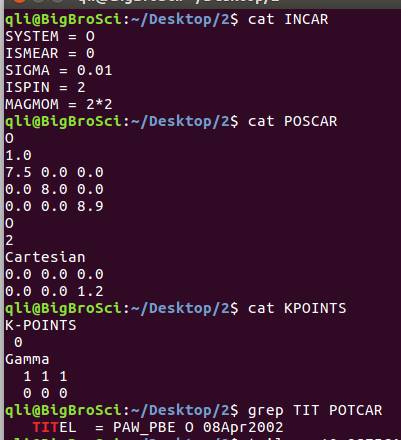
任务结束后查看OSZICAR:
从OSZICAR最后,得到体系的磁矩为2μB,你应该知道这个磁矩是怎么回事,由哪两个电子贡献。
如果不知道,看下图O$_2$的分子轨道结构:
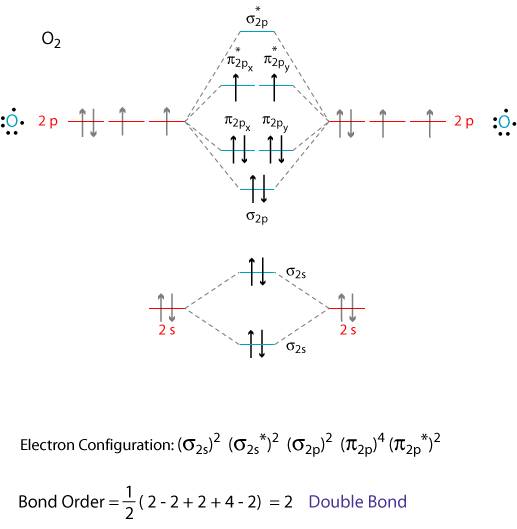
看完该图,相信大家对于O$_2$的成键方式有了一个更加深刻的印象。我们对比下VASP的输出结果。首先分析α电子的排布情况:
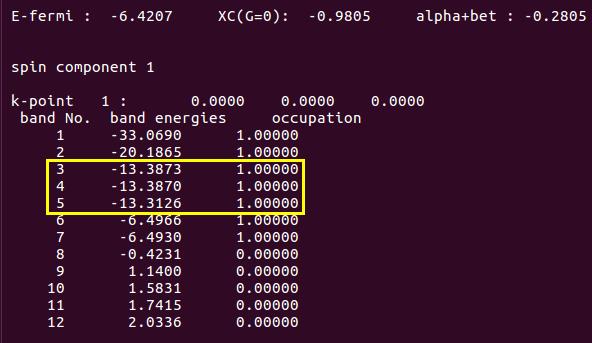
在这里,你会发现能带3和4 是简并的,应该是π(p2x)和π(p2y) 轨道中的α电子。能带 5 对应的应该是σ(2pz) 的电子。在O$_2$分子的电子构型中,两个O原子的2pz轨道以头碰头的方式形成一个σ键,其能量要比2px(2py)以肩并肩方式形成的π键能量要低。但是,能带5的能量(-13.3126)比3和4的(-13.3870)要高些,这与O$_2$的电子构型不一致,表明VASP的单点计算结果是不可靠的。
再看一下β电子的排布情况:
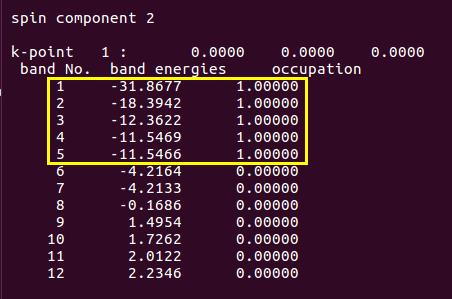
能带4和5对应的是应该是π(2px) 和π(2py) 轨道中的β电子。且总的轨道能量与前面图中一致,这说明VASP对β电子的描述是合理的。
为什么出现这样的情况呢?难道跟前面O原子的计算一样,VASP又算不准啦?
不是的,VASP怎么着也是个老牌的,响当当的计算程序,总不能让人每天指着鼻子骂算不准!
这里的主要原因是:来自于实验的键长值未必就是计算程序所认可的。也就是说,实验值和理论值之间存在偏差,实验的结构不能直接用来计算其性质,只可以作为一个理想的初始值。所以,O$_2$的分子结构需要优化一下。
VASP 优化分子结构
VASP优化分子结构的时候,需要用到一个参数:IBRION。引用官网的话:IBRION determines how the ions are updated and moved. 也就是说IBRION 这个参数决定了结构的优化过程。当你去官网查看的时候(google 搜索VASP IBRION这两个关键词),会发现IBRION有很多值。
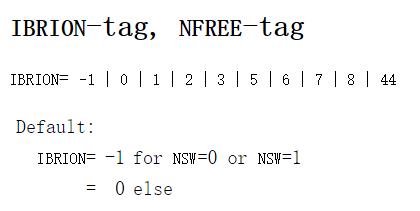
想要正确进行计算,你就需要去硬着头皮去了解各个值的含义了,这个过程必须自己去做,只听别人的建议去设置参数,而不自己去主动学习的,你的能力永远不会得到提升!!!
一般来说,优化结构的时候有3个选择:
IBRION=3:你的初始结构很差的时候;
IBRION=2:共轭梯度算法,很可靠的一个选择,一般来说用它基本没什么问题。
IBRION=1:用于小范围内稳定结构的搜索。
如果你的体系遇到结构不收敛的时候,首先检查自己的结构是否合理,也就是物理化学意义是否清晰。如果结构没问题,可以尝试下换下IBRION的参数。
下面,我们在INCAR中加上IBRION参数(IBRION=2),其他输入文件保持不变,重新进行计算:
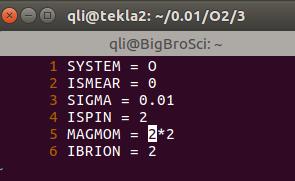
如果是用上图中的INCAR,你会发现任务很快就算完了。而且只有一步,难道输入的结构就是VASP计算出来的的稳定结构吗?有这种可能,但几率极低。
如果我们仔细查看下OUTCAR中的电子构型,发现它的信息和前面的单点计算一样。这说明,vasp并没有优化,而是又运行了一次单点计算。
为什么呢? 这是因为另一个参数:NSW。
NSW 控制几何结构优化的步数。也就是VASP进行多少离子步。
官网查看下NSW选项,发现默认值是0,也就是没有进行优化。(默认值,也叫缺省值,英文里面是 Default。 意思是,如果你不输入这个参数,程序将默认使用XXX的数值)
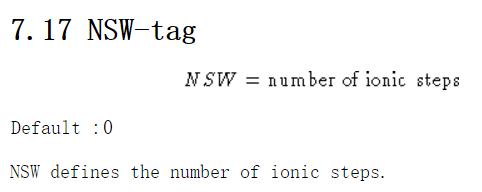
现在原因找到了,继续进行优化任务。问题来了:NSW怎么设置呢?
- 首先,它必须是大于等于0的整数。
- 其次,一般来说,简单的体系200步内就可以正常结束。
- 不知道什么时候收敛,初始结构很差,或者设置了很严格的收敛标准,那么你就要增大一下NSW的取值了,比如NSW=500或者更大。
- 我们的这个O$_2$例子很简单,设置了NSW=10(你也可以设置为100,200或者500,不会影响计算结果的。)
1 | System = O |
计算完成后,打开OSZICAR:
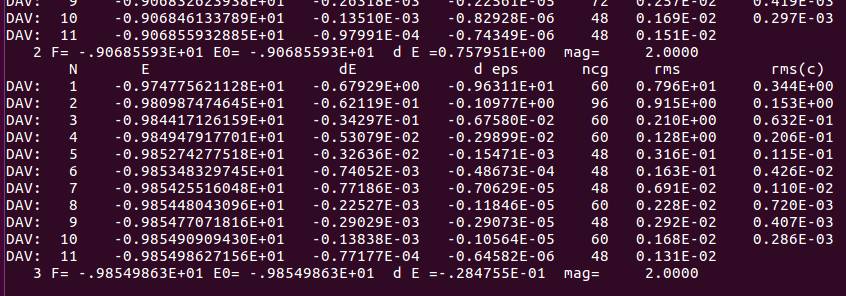
可以看到,结构优化进行了3步便停止了(如果你设置了NSW=1000,那么也是3步后就结束)。其中,每一步内又包含了若干电子步。此时的你应该知道是什么参数控制优化的结束,如果不知道请查看前面Ex09中关于收敛的文章。思考一下: 同样优化200步,设置EDIFF=1E-7和EDIFF=1E-4会有什么区别呢?
查看下OUTCAR:
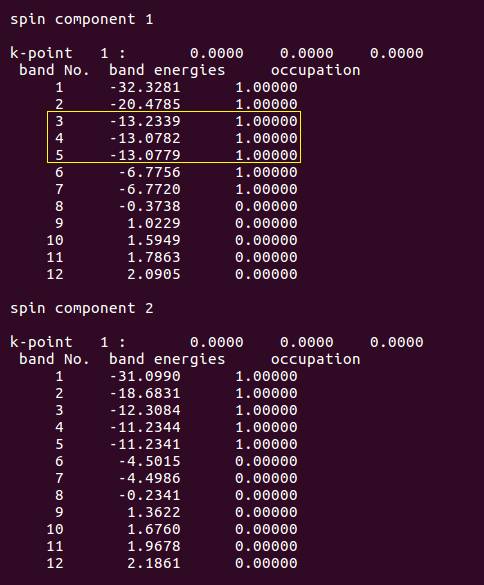
你会发现,优化过后,OUTCAR中α电子的占据状态调整过来了,β电子的保持不变。这说明计算成功了,优化起作用了。
那么优化过后的结构怎么查看呢?键长又是多少呢?
下面我们要开始真正掌握VASP的输出文件了:CONTCAR。 CONTCAR是VASP的一个输出文件,它包含了VASP计算中最后一步几何优化的结构信息,也就是优化完的结果。它也是文本格式,可以直接打开查看,如图:
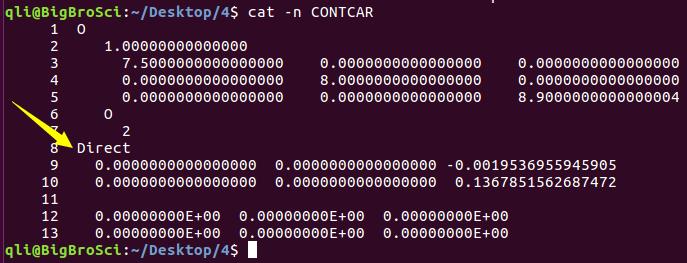
怎么才能知道优化完的O-O键长是多少呢?
1)通过坐标直接算:
此时,要注意CONTCAR输出的是Direct坐标,也就是分数坐标,需要转换成笛卡尔坐标。
xy两个方向不用考虑(都是0),z方向的坐标相减即可:键长保留四位就足够了。
(0.136785-( -0.001953))*8.9 = 1.2348 ($\AA$)
2) 使用可视化软件测量:
这样手动算,简单来说还可以,等复杂了就麻烦了,幸运的是,常用的建模软件中:p4vasp,VESTA,VMD,MS,ASE-gui等,都有测量键长的办法。这里简单讲解一下p4vasp 的用法。
p4vasp的安装
这个详见本书的附录2:https://www.bigbrosci.com/2017/11/18/A02/
1) Ubuntu 系统: sudo apt-get install p4vasp
2) Windows系统: 下载p4vasp的安装包,解压后直接打开即可。
下面是Windows下通过p4vasp读取CONTCAR的方法。Linux的也可以这样做,但终端里面:p4v CONTCAR 更高效直接。打开VASP后的界面如下: 注意图中: System:后面的三个问号部分,后面会进行对比:
点击左侧栏中的 Open 选项。下图中,左侧点击进入CONTCAR所在的目录,进入后,在右侧会显示CONTCAR
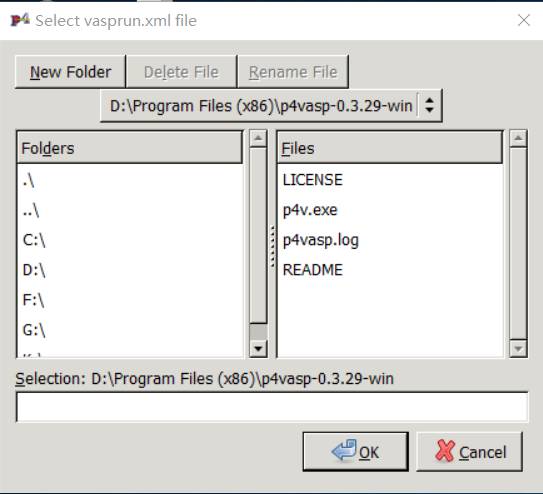
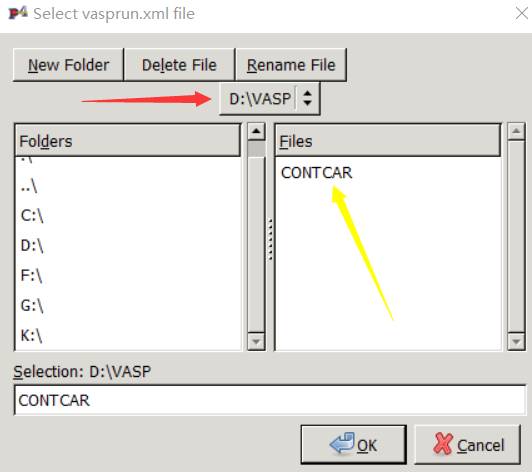
双击右侧的CONTCAR, 你会发现之前的三个问号???部分发生了变化:
显示的 O 表明VASP读取CONTCAR成功。
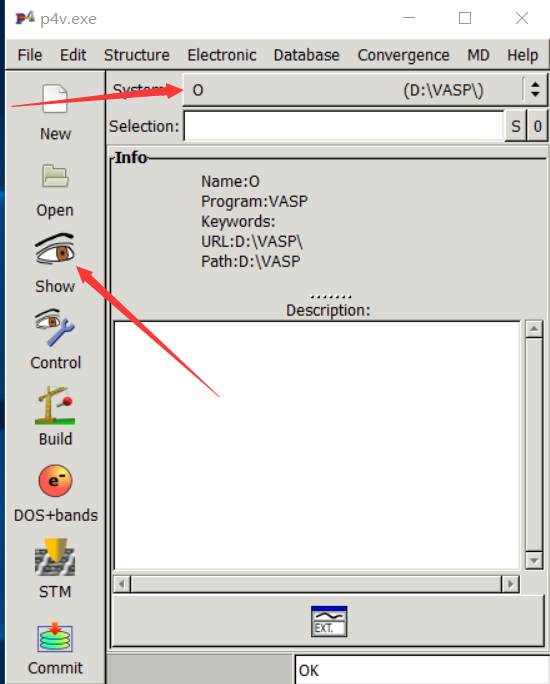
点击左侧的Show按钮,就可以查看结构了。
可视化界面的基本操作
鼠标左键按住不放,可以3维空间旋转结构;
鼠标中间摁住不放,可以上下,左右移动结构:
鼠标右键摁住不敢,动动鼠标可以缩放结构;
选择合适的观察位置:
1 把鼠标移动到你要选中的原子上,
2 通过空格(键盘上最长的键)用来选择或者取消选择原子:
3 选中两个氧原子后,如下图:
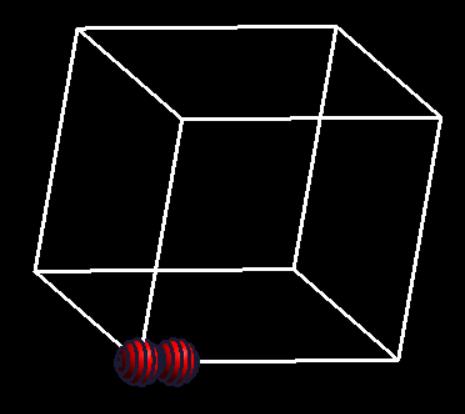
选中之后,点Structure –>Measure 显示键长为1.234774 Å,和我们手动计算的结果一样。
当然,也可以先点击:Structure –>Measure,然后再选择感兴趣的原子。
小结一下:
实验值为1.2075 Å,VASP计算结果为1.2348 Å。两者之间的差值为: 0.0289 Å,偏差为:(1.2348-1.2075)/1.2075 = 2.26%。对于理论和实验之间的偏差,如果小于5%,我们一般可以认为吻合的很好。有时候很多同学揪着VASP的计算结果与实验值的偏差不放,误差已经千分之几了,感觉心里还是不放心,有着一种不完全匹配不罢休的冲动。这大可不必放在心上,如果你的结果偏离实验值千分之几,直接用就可以了。
“To err is human; to describe the error properly is sublime.”
— Cliff Swartz, Physics Today 37 (1999),388.
对于其他的软件程序(MS,VESTA,VMD等),大家下载安装后,百度里面搜一搜教程,基本操作应该很快就能掌握。
扩展练习:
1 IBRION:https://cms.mpi.univie.ac.at/vasp/vasp/IBRION_tag_NFREE_tag.html
2 NSW:http://cms.mpi.univie.ac.at/vasp/guide/node108.html
3 CONTCAR:https://cms.mpi.univie.ac.at/wiki/index.php/CONTCAR
4 从头开始重现本节的所有操作;
5 尝试不同的初始键长,运行vasp,查看输出结果。
总结:
1)学会结合自己所学的化学知识,分析双原子的电子构型;
2)知道IBRION + NSW进行结构优化;
3)知道什么参数控制结构优化的停止,以及单个离子步内电子步数;
4)学会使用可视化软件查看输出的几何结构;
5)知道理论结果和实验值之间没有100%吻合。